LAMMPS 代表 Large-scale Atomic/Molecular Massively Parallel Simulator。
LAMMPS 是一种经典的分子动力学仿真代码,专注于材料建模。它旨在在并行计算机上高效运行,并且易于扩展和修改。LAMMPS 最初由美国能源部机构桑迪亚国家实验室开发,现在包括来自许多机构的许多研究小组和个人的贡献。LAMMPS 的大部分资金来自美国能源部 (DOE)。LAMMPS 是根据 GNU 公共许可证版本 2 (GPLv2) 的条款分发的开源软件。
本教程将通过 3 种不同的方式:velocity、fix move、fix setforce 分别移动 3 个原子
- 3 种原子运动控制方式(velocity、fix move、fix setforce)的具体设置
- 模拟系统的基本参数(边界、区域、单位等)如何定义
- 输出文件的配置方式
- 各原子运动规律的理论依据
教程链接: go.openbayes.com/GdLQk
该教程将在云平台 OpenBayes.com 上使用 RTX 4090 进行演示,使用下方邀请链接注册即可获得 4 小时 RTX 4090 免费使用时长:
一、输入文件说明
in.txt 输入文件
文件解析
csharp
#3 个原子运动演示
#设置模拟单位系统为 Lennard-Jones(LJ)单位
#LJ 单位是分子模拟中常用的无量纲单位,便于不同体系间的比较
units lj
#设置原子样式为atomic(仅包含原子类型和位置信息,无分子结构)
atom_style atomic
#定义边界条件:3 个方向均为固定边界(f = fixed)
#固定边界意味着原子无法穿过边界,也不会从另一侧周期性出现
boundary f f f
#定义模拟区域的几何范围
#格式:region <区域名> block <x起始> <x结束> <y起始> <y结束> <z起始> <z结束>
#此处创建一个名为「box」的长方体区域,X 方向 0-150,Y 方向 0-300,Z 方向 0-10
region box block 0 150 0 300 0 10
#基于定义的区域创建模拟盒子
#格式:create_box <原子类型数量> <区域名>
#此处创建包含1种原子类型的模拟盒子,边界由"box"区域定义
create_box 1 box
#在模拟盒子中创建 3 个原子(均为类型1)
#格式:create_atoms <原子类型> single <x坐标> <y坐标> <z坐标>
#3 个原子初始 Y 坐标均为 10.0,X 坐标分别为 30.0、70.0、110.0(横向分开便于观察)
create_atoms 1 single 30.0 10.0 5.0 # ID=1(用于演示 velocity 命令)
create_atoms 1 single 70.0 10.0 5.0 # ID=2(用于演示 fix move 命令)
create_atoms 1 single 110.0 10.0 5.0 # ID=3(用于演示 fix setforce 命令)
#设置原子类型 1 的质量为 1.0(LJ 单位下)
#质量会影响由 fix setforce 产生的加速度(a = F/m)
mass 1 1.0
#设置原子间相互作用势类型为 none(无相互作用)
#关闭原子间作用力,避免相互干扰,使每种运动方式的效果更纯粹
pair_style none ---------------- 原子 1:使用velocity命令控制运动 ----------------
bash
#1. 创建原子组vel_group,仅包含ID=1的原子
group vel_group id 1
#2. 使用 velocity 命令设置原子的初始速度
#格式:velocity <组名> set <vx> <vy> <vz>
#此处设置 X 和 Z 方向速度为 0.0,Y 方向速度为 50.0(沿 Y 轴正方向运动)
#velocity 命令设置的速度会通过后续的积分器更新位置,保持匀速运动(无外力时)
velocity vel_group set 0.0 50.0 0.0 ---------------- 原子 2:使用 fix move 命令控制运动 ----------------
bash
#1. 创建原子组 move_group,仅包含 ID=2 的原子
group move_group id 2
#2. 先将原子初始速度设为 0.0(确保初始静止)
velocity move_group set 0.0 0.0 0.0
#3. 使用 fix move 命令强制原子运动
#格式:fix <ID> <组名> move linear <vx> <vy> <vz>
#此处设置 X 和 Z 方向速度为 0.0,Y 方向强制速度为 30.0
#fix move 命令会直接按指定速度更新位置,不受动力学方程影响(强制匀速)
fix 2 move_group move linear 0.0 30.0 0.0 ---------------- 原子 3:使用 fix setforce 命令控制运动 ----------------
python
创建原子组 force_group,仅包含 ID=3 的原子
group force_group id 3
#2. 先将原子初始速度设为 0.0(确保初始静止)
velocity force_group set 0.0 0.0 0.0
#3. 使用 fix setforce 命令施加恒定力
#格式:fix <ID> <组名> setforce <fx> <fy> <fz>
#此处设置X和Z方向力为 0.0,Y 方向力为 20.0
#力会产生加速度(a=F/m=20.0/1.0=20.0),使原子做匀加速运动(速度随时间增加)
fix 3 force_group setforce 0.0 20.0 0.0
#添加时间积分器,用于更新原子位置和速度
#fix nve 表示使用 NVE 系综(微正则系综),守恒粒子数、体积和能量
#积分器会根据牛顿运动方程,结合速度、力等参数计算每一步的位置变化
fix 1 all nve
#设置轨迹输出参数
#1. dump 命令:每1步记录一次所有原子的位置信息,输出到 trajectory.xyz 文件
#格式:dump <ID> <组名> atom <输出频率> <文件名>
dump 1 all atom 1 trajectory.xyz
#2. 调整轨迹文件格式:按原子 ID 排序输出,使结果更易读
dump_modify 1 sort id
#设置模拟时间参数
#1. 设置时间步长为 0.01(LJ 单位下的时间)
#时间步长是模拟中最小的时间单位,决定计算精度和效率
timestep 0.01
#2. 设置热力学信息输出频率:每 10 步输出一次
thermo 10
#3. 设置热力学输出内容:步数、温度、动能、势能、总能量
thermo_style custom step temp ke pe etotal
#运行模拟:总步数为 500 步
#总模拟时间 = 步数 × 时间步长 = 500 × 0.01 = 5.0(LJ 时间单位)
run 500
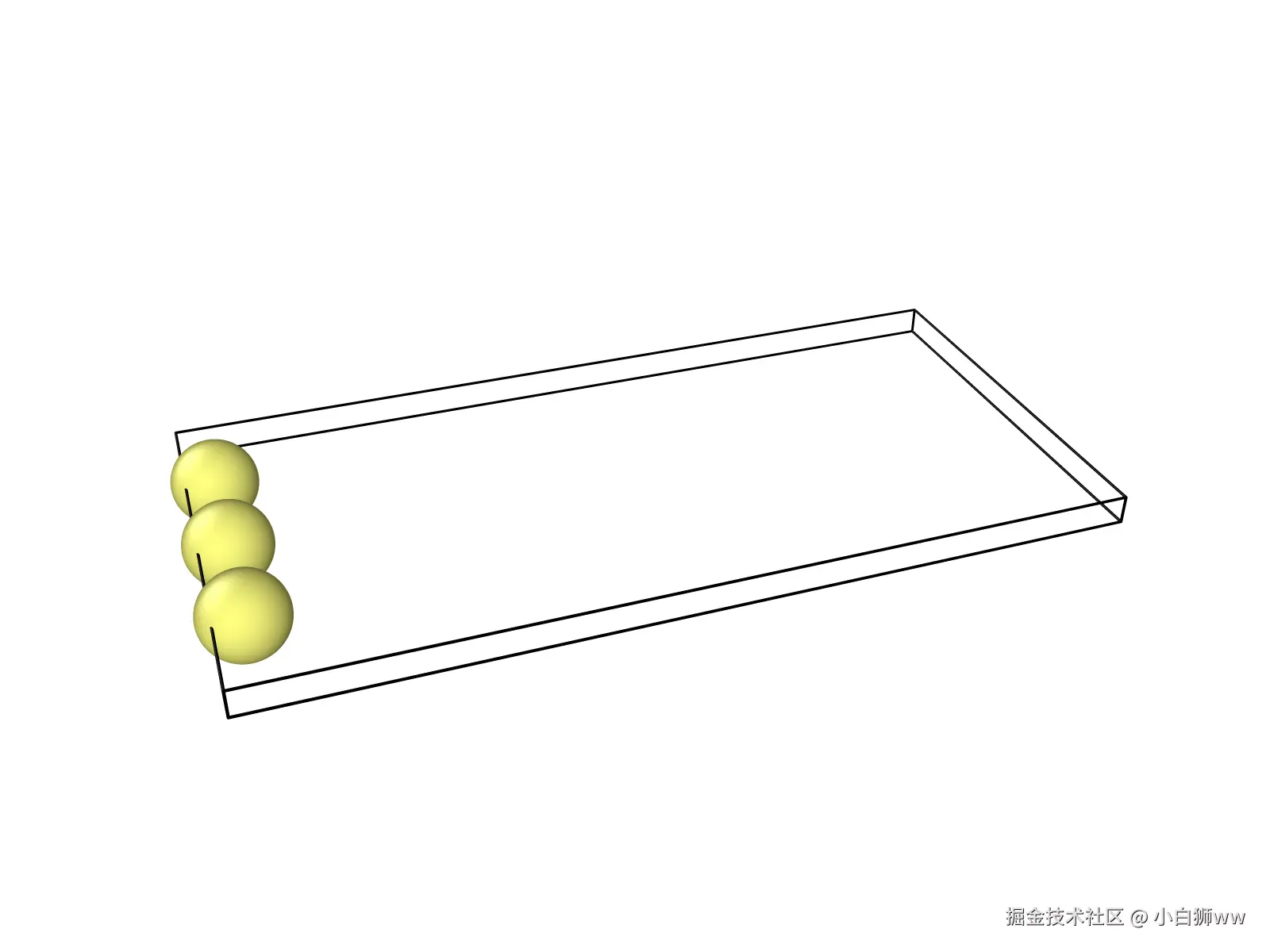
#各原子 Y 方向运动预期结果(初始 Y=10.0):
#- 原子 1(velocity):匀速运动,位移=速度×时间=50.0×5.0=250.0 → 最终 Y≈260.0
#- 原子 2(fix move):强制匀速,位移=强制速度×时间=30.0×5.0=150.0 → 最终 Y≈160.0
#- 原子 3(fix setforce):匀加速运动,位移=0.5×加速度×时间²=0.5×20.0×5.0²=250.0 → 最终 Y≈260.0二、操作步骤
- 克隆并启动容器
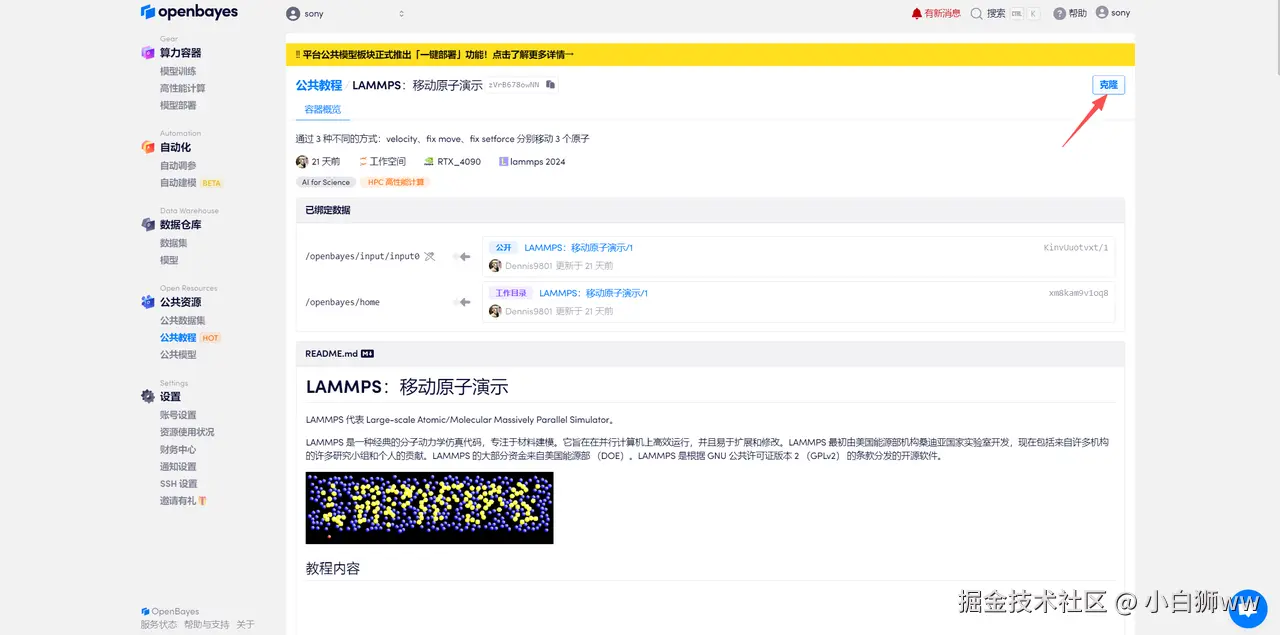
登录 OpenBayes.com,在「公共教程」页面,点击「HPC 高性能计算」栏目,选择「LAMMPS:移动原子演示」教程。
 页面跳转后,点击右上角「克隆」,将该教程克隆至自己的容器中。
页面跳转后,点击右上角「克隆」,将该教程克隆至自己的容器中。
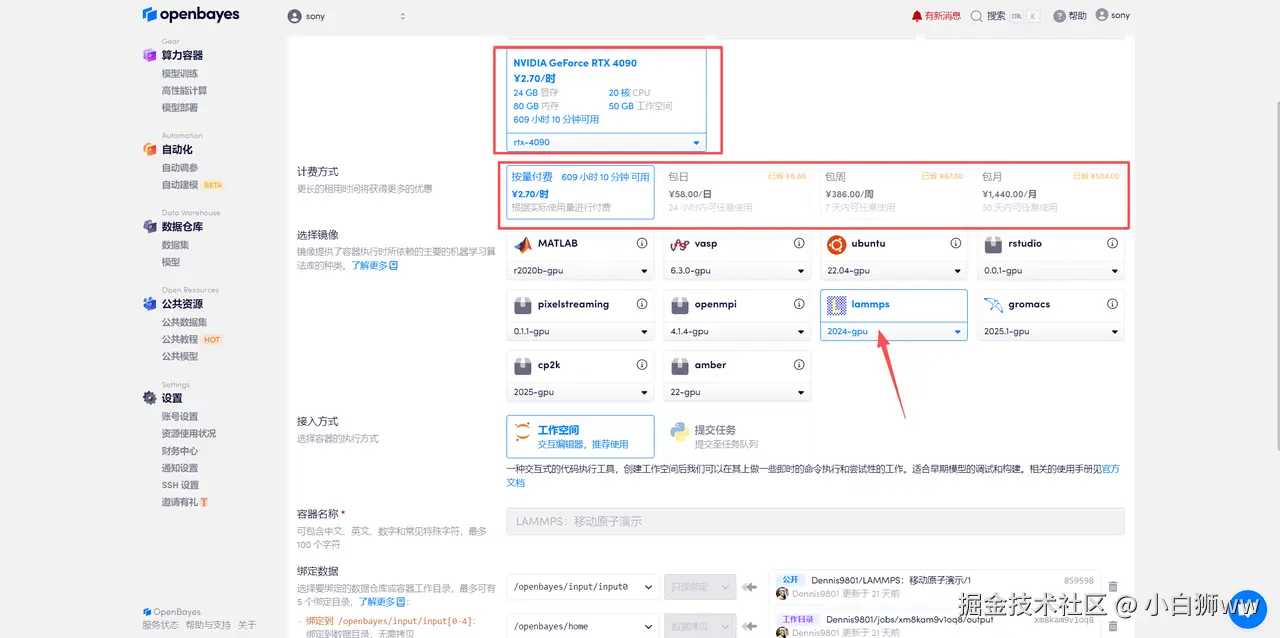
 选择「NVIDIA GeForce RTX 4090」以及「lammps」镜像,OpenBayes 平台提供了 4 种计费方式,大家可以按照需求选择「按量付费」或「包日/周/月」,点击「继续执行」。可以使用下面的邀请链接,获得 RTX 4090 使用时长!
选择「NVIDIA GeForce RTX 4090」以及「lammps」镜像,OpenBayes 平台提供了 4 种计费方式,大家可以按照需求选择「按量付费」或「包日/周/月」,点击「继续执行」。可以使用下面的邀请链接,获得 RTX 4090 使用时长!
openbayes.com/console/sig...
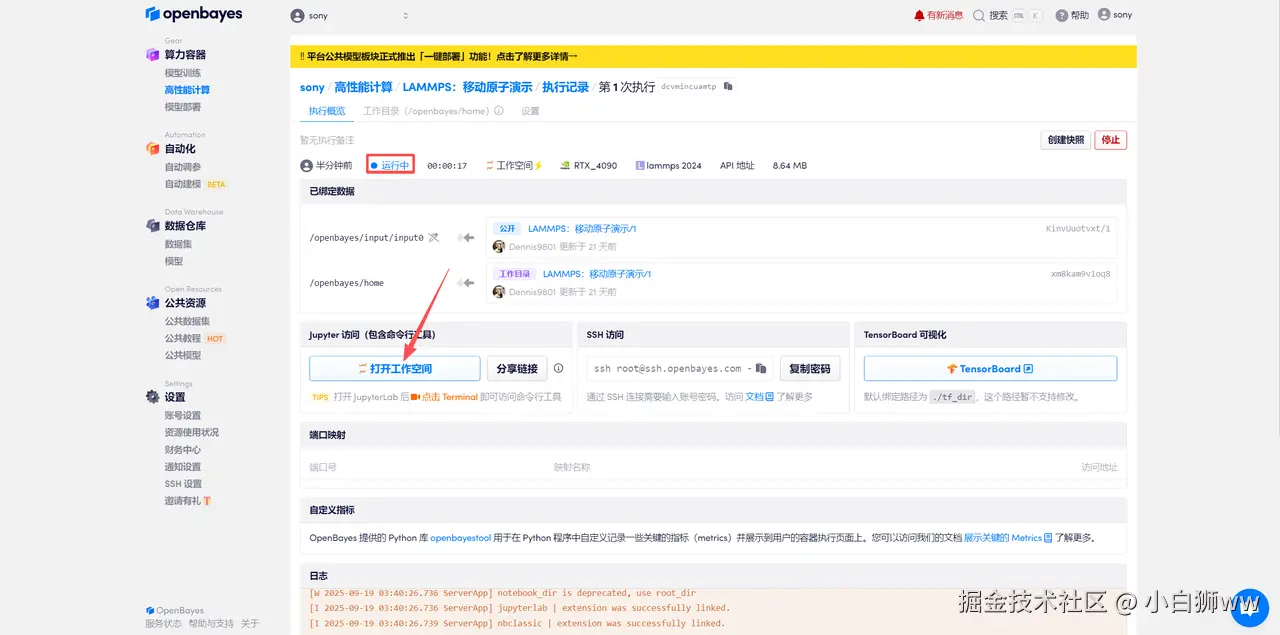
待系统分配好资源,当状态变为「运行中」后,点击「打开工作空间」。
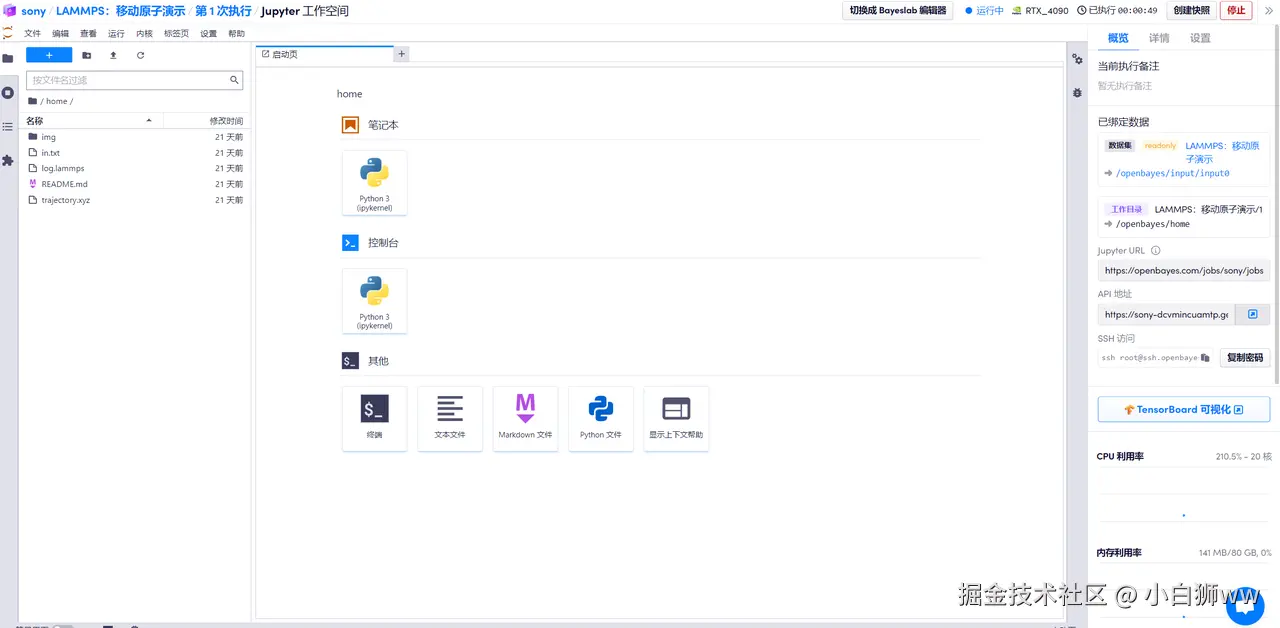 2. 运行 lammps
2. 运行 lammps
首先打开「终端」,输入以下命令运行 lammps。
arduino
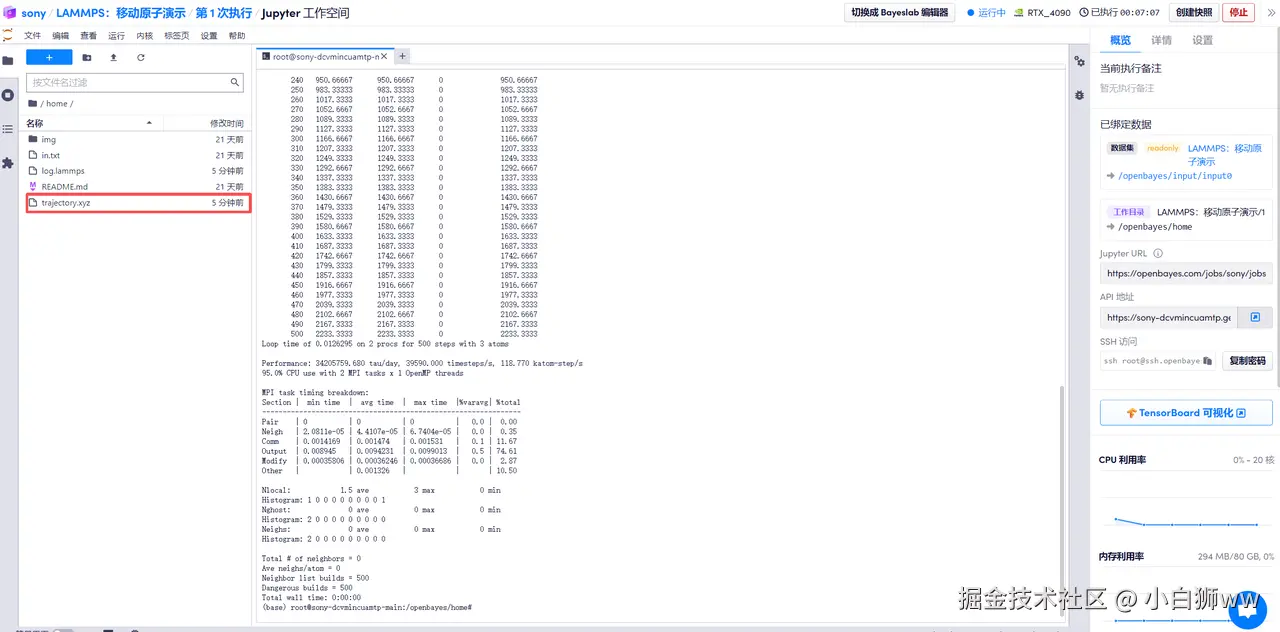
mpirun --allow-run-as-root -np 2 lmp -sf gpu -pk gpu 1 < in.txt  3. 文件输出
3. 文件输出
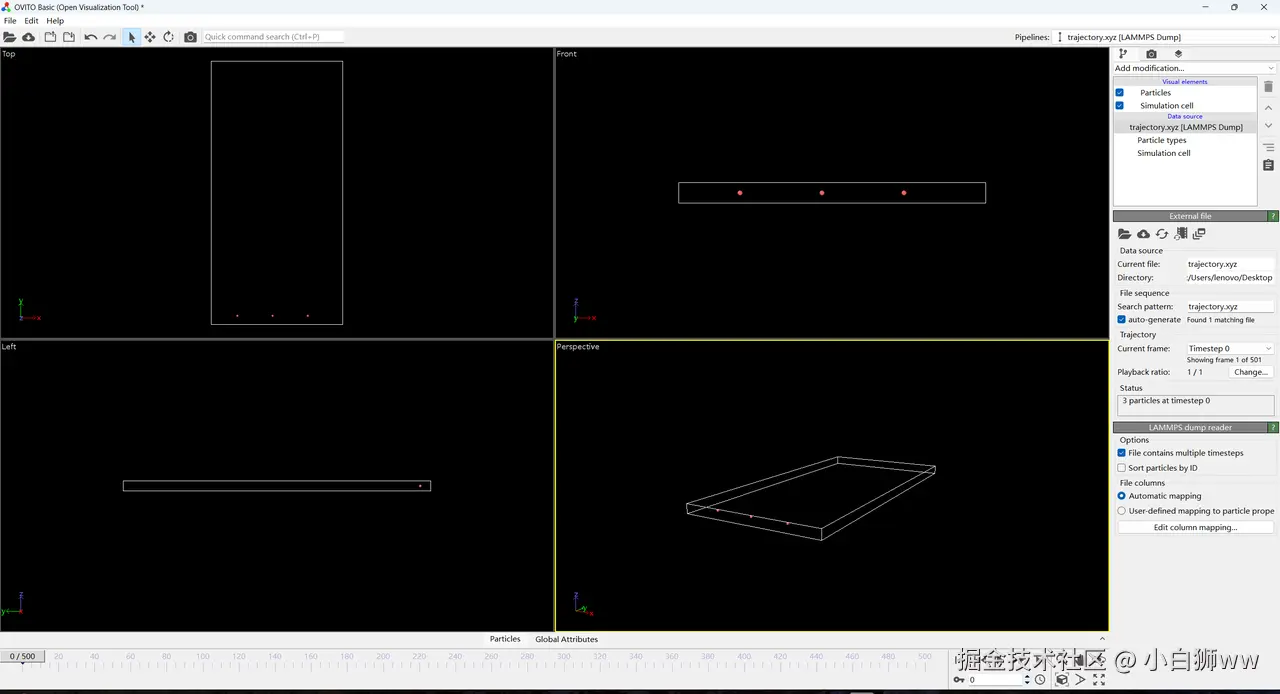
待模型运行完成后,得到一个输出文件「trajectory.xyz」,这是原子轨迹文件,可通过 VMD、Ovito 等软件可视化拉伸过程中的晶体结构演变。我们将该文件下载到本地并载入到 Ovito 软件中,即可得到原子运动模拟视图。