我们通过R语言下载TCGA数据时需要导入TCGAbiolinks包的,但是这个无法直接导入,需要我们先导入BiocManager包
1.1****导入 BiocManager
|--------------------------------------------------------|
| R install.packages("BiocManager") library(BiocManager) |
1.2****导入 TCGA 数据下载所需的包
|-----------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------|
| C++ BiocManager::install("remotes") BiocManager::install("BioinformaticsFMRP/TCGAbiolinksGUI.data") BiocManager::install("ExperimentHub") BiocManager::install("BioinformaticsFMRP/TCGAbiolinks") library(TCGAbiolinks)#加载包 |
这个需要先安装前三个才可以安装第四个,同时非常难安装!!!
各种癌性缩写的网站
https://www.jianshu.com/p/3c0f74e85825
固定的格式
|---------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------|
| cancer_type = "TCGA-LUAD" #肿瘤类型,这里可修改癌症类型 #TCGA 肿瘤缩写:https://www.jianshu.com/p/3c0f74e85825 expquery <- GDCquery(project = cancer_type, data.category = "Transcriptome Profiling", data.type = "Gene Expression Quantification", workflow.type = "STAR - Counts" ) GDCdownload(expquery,directory = "GDCdata") expquery2 <- GDCprepare(expquery,directory = "GDCdata",summarizedExperiment = T) save(expquery2,file = "luad.gdc_2022.rda") # 保存 rda格式 |
由于文件较大,我采取直接导入文件的方式
导入文件后,重新打开Rstudio,一定要转到TCGAdata目录下
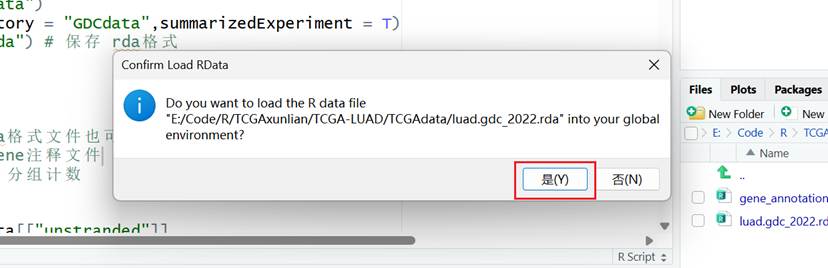
方式一:
双击文件夹中的文件,在跳出的提示中点击是
方式二:
注: 这个需要在文件的目录下进行,否则会报错
|-----------------------------|
| R load("luad.gdc_2022.rda") |
2.1****数据处理
2.1.1****导入文件
|------------------------------------|
| R load("gene_annotation_2022.rda") |
2.1.2****分组计数 table
|------------------------------------|
| R table(gene_annotation_2022$type) |
$ 是提取数据表中的列名,这句代码表达的是提取gene_annotation_2022.rda中的type列,并且输出type列的值以及对应的数量
|--------------------------------------------------------------------------------------------------------------------|
| 在TCGA中,基因表达谱有多种形式,例如 counts,tpms等格式 * counts 只用来做差异分析,差异分析也只能用counts来做---针对TCGA数据库 * tpms 除了差异分析,其他都用tpms |
2.2.3****提取 counts
counts里面都是整数
无需理解,记住,每个都一样
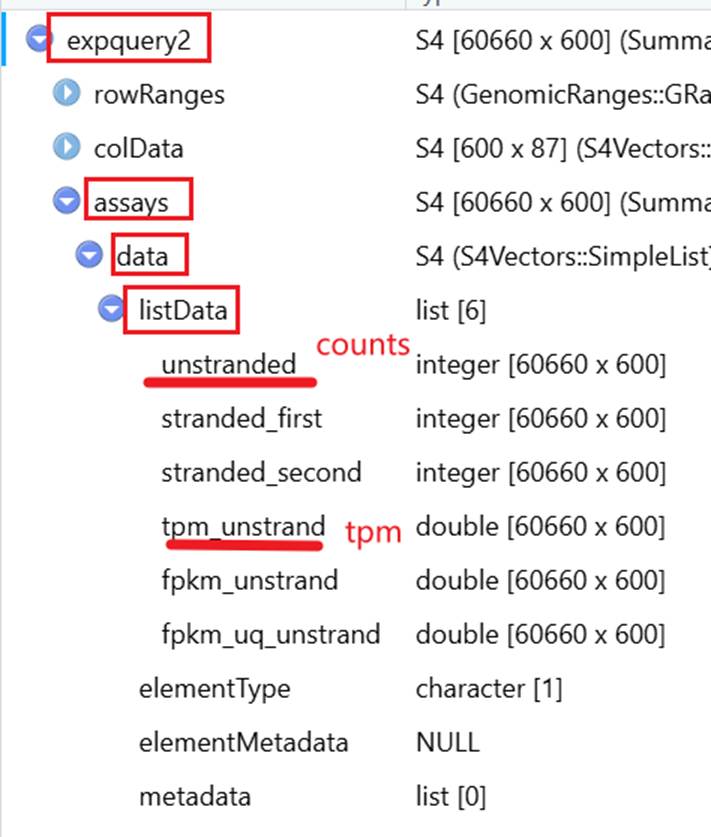
|-----------------------------------------------------------------------------------------------------------------------------------------------------------------------------------|
| R counts <- expquery2@assays@data@listData\["unstranded"] colnames(counts) <- expquery2@colData@rownames #给予列名 rownames(counts) <- expquery2@rowRanges@ranges@NAMES #给予行名 |
expquery2 是我们下载的数据并处理的名字
2.2.4****基因 ID 转换
我们要将counts中行名从ENSEMBL转为symbol
把ID值改为名字
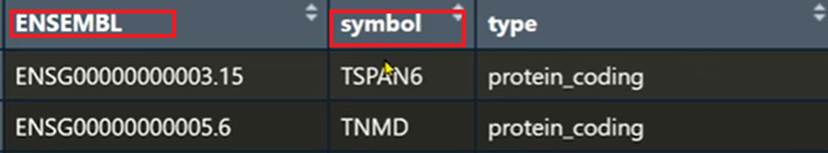
我们先逐步来看
|----------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------|
| R #同上 counts1 <- expquery2@assays@data@listData\["unstranded"] colnames(counts1) <- expquery2@colData@rowname rownames(counts1) <- expquery2@rowRanges@ranges@NAMES counts1 <- as.data.frame(counts1) #此时的counts1还不是数据表的形式,这句把其变为数据表 #将counts1 的行名变为一列,列名为 ENSEMBL counts1 <- rownames_to_column(counts1,var='ENSEMBL') #两个表按照 ENSEMBL 列进行合并,效果为在counts1中,多了symbol 和 type 列 counts1 <- inner_join(counts1,gene_annotation_2022,"ENSEMBL") #因为是二维数据表,所以要加逗号 #按照symbol列的每行进行去重复 counts1 <- counts1!duplicated(counts1$symbol), |
使用传导符
|----------------------------------------------------------------------------------------------------------------------------------------------------------------------------------|
| R counts <- counts %>% as.data.frame() %>% rownames_to_column("ENSEMBL") %>% inner_join(gene_annotation_2022,"ENSEMBL") %>% .!duplicated(.$symbol), #这里的 . 是为了告诉传导符传到哪里 |
2.2.5****比较两个表是否相同 identical
我们上面产生了counts和counts1两个表,要比较它俩是否完全一致(包括顺序),需要用到identical
示例:
|-----------------------------------------------------------------------------------------------|
| JSON a <- c("a","b","a","b","c") b <- c("a","b","b","a","c") identical(a,b) #输出 1 FALSE |
|-----------------------------------------------------------------------------------------------|
| R identical(colnames(counts),colnames(counts1)) identical(rownames(counts),rownames(counts1)) |
2.2.6****更改行名
此时,我们的行名如下图,我们要将行名改为基因名,前面我们已经对 symbol 列进行去重(这是因为R语言要求行名不可以重复),现在我们就是将列转为行。
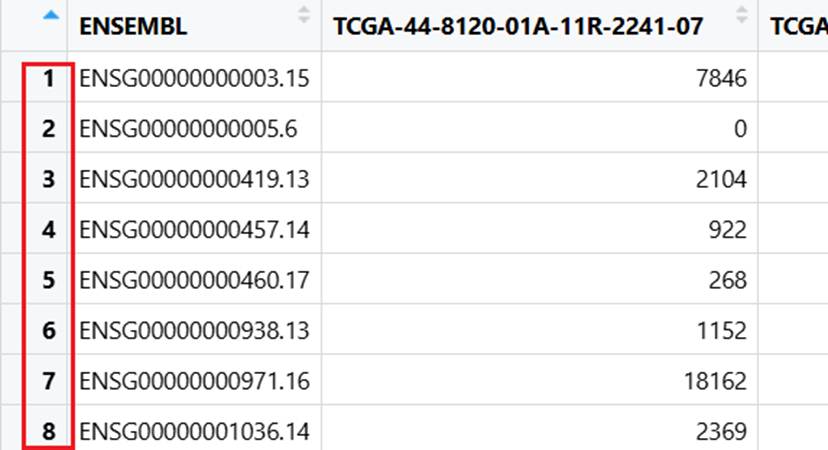
|--------------------------------------------------------------------------------------------------------------------------------------------------------------------------|
| R rownames(counts) <- NULL counts <- counts %>% column_to_rownames("symbol") #拆解 rownames(counts1) <- NULL #无 counts1 <- column_to_rownames(counts1,var = 'symbol') |
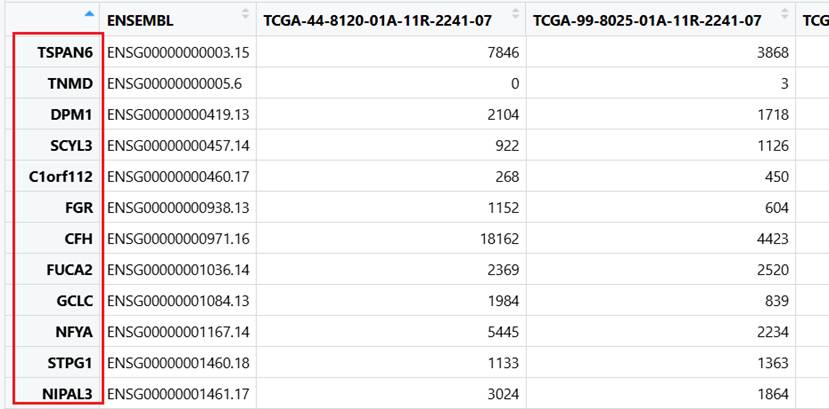
2.2.7****筛选基因
我们基因类型有很多,当我们研究特定类型的基因时就需要进行筛选,方法如下:
以筛选 protein_coding 类型为例:
|------------------------------------------------------------------------------------------------------------|
| R table(countstype)#(注:可通过table(countstype)查看基因类型) counts <- countscounts$type == "protein_coding", |
2.2.8****删除无关列
在count表中,我们当时为添加symbol ,type等列,将原本的行名改为列名,现在已经不需要ENSEMBL,type 等列,它们恰好在第一列和最后一列,我们删除这两列
|---------------------------------------------------------|
| R ncol(counts) #counts 表有多少列 nrow(counts) #counts 表有多少行 |
|--------------------------------------------|
| R counts <- counts,-c(1,ncol(counts)) |
2.2.9****对列处理
- 将列名切割为16位字符,同时去重复
|-------------------------------------------------------------------------------------------------------------|
| R colnames(counts) <- substring(colnames(counts),1,16) counts <- counts,!duplicated(colnames(counts)) |
- 保留特定后三位列
01A 肿瘤样本 11A 正常样本
|-----------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------|
| R # 保留01A (注:可通过table(substring(colnames(counts),14,16))查看样本类型) counts01A <- counts,substring(colnames(counts),14,16) == c("01A") # 保留11A counts11A <- counts,substring(colnames(counts),14,16) == c("11A") |
2.2.10****提取 tpms
|-----------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------|
| SQL #和counts基本一模一样 tpms <- expquery2@assays@data@listData\["tpm_unstrand"] colnames(tpms) <- expquery2@colData@rownames rownames(tpms) <- expquery2@rowRanges@ranges@NAMES tpms <- tpms %>% as.data.frame() %>% rownames_to_column("ENSEMBL") %>% inner_join(gene_annotation_2022,"ENSEMBL") %>% .!duplicated(.$symbol), rownames(tpms) <- NULL tpms <- tpms %>% column_to_rownames("symbol") # 保留mRNA (注:可通过table(tpmstype)查看基因类型) tpms \<- tpms\[tpmstype == "protein_coding",] tpms <- tpms,-c(1,ncol(tpms)) # 把TCGA barcode切割为16位字符,并去除重复样本 colnames(tpms) <- substring(colnames(tpms),1,16) tpms <- tpms,!duplicated(colnames(tpms)) # 保留01A (注:可通过table(substring(colnames(tpms),14,16))查看样本类型) tpms01A <- tpms,substring(colnames(tpms),14,16) == c("01A") # 保留11A tpms11A <- tpms,substring(colnames(tpms),14,16) == c("11A") |
2.2.11****保存数据
|-------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------|
| R write.table(counts01A,"counts01A.txt",sep = "\t",row.names = T,col.names = NA,quote = F) write.table(counts11A,"counts11A.txt",sep = "\t",row.names = T,col.names = NA,quote = F) write.table(tpms01A,"tpms01A.txt",sep = "\t",row.names = T,col.names = NA,quote = F) write.table(tpms11A,"tpms11A.txt",sep = "\t",row.names = T,col.names = NA,quote = F) |
2.2.12****按列合并,按行合并
- 按列合并: 行名要求一致,把每个表除行名外的列进行合并
在合并之前一定要检查行名是否一致!!!
|----------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------|
| R # cbind 按列合并 counts <- cbind(counts01A,counts11A) tpms <- cbind(tpms01A,tpms11A) write.table(counts,"counts.txt",sep = "\t",row.names = T,col.names = NA,quote = F) write.table(tpms,"tpms.txt",sep = "\t",row.names = T,col.names = NA,quote = F) |
- 按行合并:列名要求一致,把每个表除列名外的行进行合并
在合并之前一定要检查列名是否一致!!!
2.2.13****对表数据进行转化处理
先看一下表格中数据的最大值和最小值
|------------------------------------------------------------------------------------------------------------------|
| R > range(tpms)#查看数据范围 1 0.0 136166.7 > range(tpms01A) 1 0.0 136166.7 > range(tpms11A) 1 0 106178 |
我们发现数字的差距很大,所以要对其中的数据进行缩小,采取的是log2转化
后续我们都是使用转化后的数据来进行处理
|---------------------------------------------------------------------------------------------------------------------------------------------------------------------------------|
| R tpms_log2 <- log2(tpms+1)#log2转换 为什么要加1,因为因变量不能为0 range(tpms_log2) tpms01A_log2 <- log2(tpms01A+1) range(tpms01A_log2) tpms11A_log2 <- log2(tpms11A+1) range(tpms11A_log2) |
结果:
|------------------------------------------------------------------------------------------------------------------------------------------|
| R > range(tpms_log2) 1 0.00000 17.05503 > range(tpms01A_log2) 1 0.00000 17.05503 > range(tpms11A_log2) 1 0.00000 16.69614 |
保存
|---------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------|
| R write.table(tpms_log2,"tpms_log2.txt",sep = "\t",row.names = T,col.names = NA,quote = F) write.table(tpms01A_log2,"tpms01A_log2.txt",sep = "\t",row.names = T,col.names = NA,quote = F) write.table(tpms11A_log2,"tpms11A_log2.txt",sep = "\t",row.names = T,col.names = NA,quote = F) |