------科研人的效率革命,从这一刻开始
大模型已经帮助科研的每一个环节:文献管理、代码调试、实验设计、模型构建...... 但科研绘图依然是多数研究生头疼的环节之一:
-
MATLAB / Python 的语法太繁琐
-
ggplot2 语法记不住
-
论文级图形需要反复调试
-
一个图可能要改几十遍
💡 而现在,Gemini 3(Google 最新大模型)+ 服务器算力正在让科研绘图这件事变得轻松、高效、甚至准一键分析。
先产生作图代码,再调用程序跑
什么是 Gemini 3?
Gemini 3 是 Google 最新一代的通用人工智能大模型,也是当前最强的科研辅助模型之一。在以下核心能力上实现了质的飞跃:
更强的多模态理解能力(文本 + 图像 + 数据)
Gemini 3 可以同时理解论文、图像、表格、代码、实验数据,因此特别适合科研---它能像研究生一样读图、读文献、理解实验流程、解释图像结构和推导逻辑。
极强的长上下文能力(百万级 Token)
可一次输入整份论文集、整个项目的代码仓库、上万条数据文件,而不需要拆分上传。
强大的代码生成能力(Python / R / MATLAB / Bash)
Gemini 3 能生成直接可运行的科研代码,而且 debug 能力非常强,适合生信工作流:RNA-seq → ChIP-seq → ATAC-seq → WGS → 统计建模 → 可视化
支持持续对话式科研
你可以像带一个助理一样不断提出修改要求,它会自动优化脚本、调整绘图参数、修复报错、补全分析流程。
更高的科学推理能力
面对数学、信号处理、统计学、生物学机制推理问题,Gemini 3 的链式推理能力更准确、逻辑性更强,能够做严格的科研解释与模型搭建。
Gemini 3 对生信领域有什么益处?
成倍提升分析速度
Gemini 3 能极大减少重复性操作,适用于:
-
ChIP-seq / ATAC-seq / RNA-seq 全流程脚本自动生成
-
自动写 Snakemake / Nextflow pipeline
-
自动 QC 报告生成(FastQC、MultiQC)
-
自动找 bug,自动补齐缺失步骤
你只需一句话:
帮我用 deepTools 画一个 H3K27ac peak 的热图。
它就能接管整个过程。
科研绘图达到 SCI 级别
Gemini 3 能准确分析矩阵数据、样本分组、基因注释,自动生成标准的 SCI 图:(逻辑是先生成绘图代码,再运行代码生成图片)
-
火山图、热图
-
UMAP/t-SNE/PCA
-
GO/KEGG 富集气泡图
-
ChIP-seq peaks 位置分布图
-
bulk RNA-seq 基因表达趋势图
-
motif 分析结果示意图
省大量人力成本
传统生信分析往往需要:
⏳ 解决报错 ⏳ 修改 R 脚本 ⏳ 调整图形 ⏳ 反复阅读参数文档 ⏳ 在服务器找文件路径
Gemini 3 会自动完成这些工作,让你把时间专注在科学问题本身
结合服务器算力直接跑大数据
Gemini 3 + 服务器环境可以做到:
-
直接从服务器目录读取 10GB~TB 级数据
-
自动并行加速
-
自动把 Python/R 代码推上 GPU/CPU
-
自动保存高分辨率图(PDF/SVG/PNG)
适用于:
-
单细胞 10X 大矩阵
-
数百样本 bulk RNA-seq
-
大规模组学数据
-
分子动力学模拟结果
-
ATAC-seq / ChIP-seq 轨迹图和 bigWig 文件分析
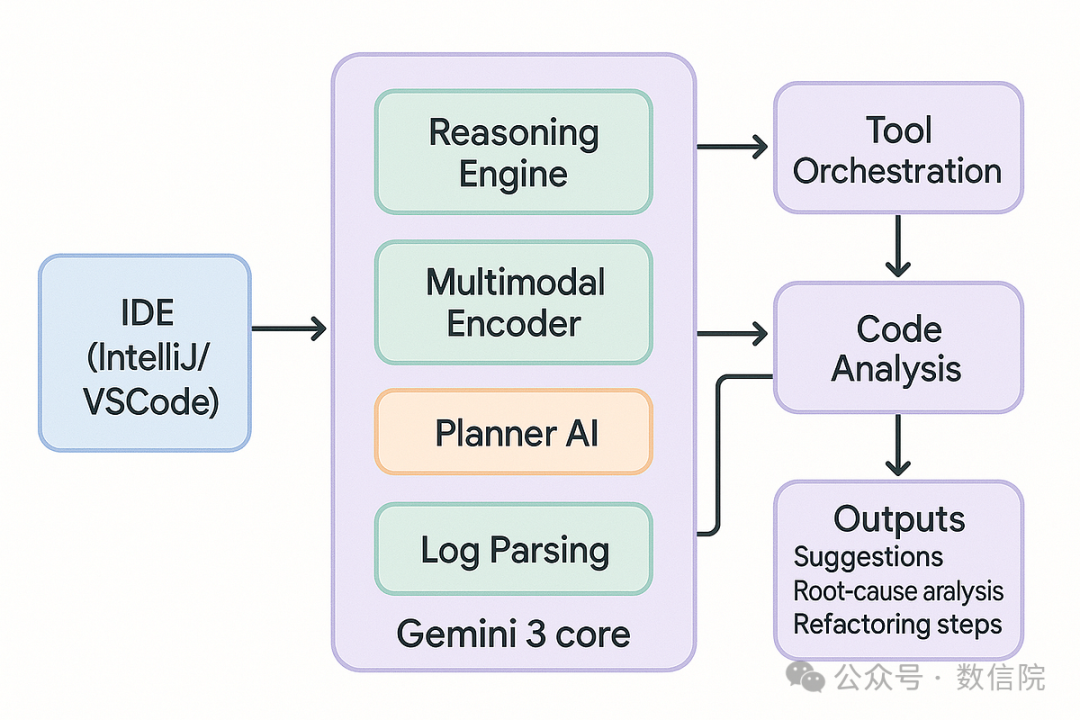
为什么要在服务器上使用 Gemini 3 做分析?
Image
与在本地电脑运行相比,把 Gemini 3 部署到服务器上,你能获得:
1.更强算力,轻松处理大规模数据
服务器的 CPU/GPU 能力远超本地笔电,适合:
-
大样本队列
-
单细胞、多组学大矩阵
-
大型材料模拟数据
- 高质量论文级绘图自动生成
你只需要一句话:
帮我画出差异基因火山图,SCI 风格。 Gemini 3 可以自动:
-
读取服务器文件
-
分析数据
-
生成 Python 或 matplotlib 或 R代码
-
自动调色、字号、线宽、标注
-
保存矢量图(PDF/SVG)
3.模型长对话能力强,可连续优化图形
你可以不断调整:
标签再小一点
去掉网格线,增加字体
标注不同的marker
Gemini 3 会自动更新代码,无需你手动 debug。
环境准备:如何在服务器上启用 Gemini 3
这一部分详情参考:
https://mp.weixin.qq.com/s/aqBQ95MHCxCYid9j_fkjCg
用 Gemini 3 生成 SCI 级别绘图的全过程演示
以下展示真实科研场景:绘制差异基因火山图。
Step 1:你上传需求 + 数据路径
例如你告诉 Gemini:
当前目录下05.DE是差异分析的结果,帮我根据这个数据画一个sci级别的火山图
Step 2:Gemini 自动生成分析代码
下面是 Gemini 3 会自动写出的 高质量可运行代码:
import pandas as pd
import numpy as np
import matplotlib.pyplot as plt
import seaborn as sns
def plot_sci_volcano(file_path, output_name='volcano_plot'):
# 1. 读取数据
try:
df = pd.read_csv(file_path, sep='\t')
except FileNotFoundError:
print(f"错误:找不到文件 {file_path}")
return
# 2. 检查列名
col_map = {
'logfc': 'log2FoldChange',
'padj': 'padj',
'gene': 'gene_id'
}
# 简单的列名自动匹配尝试
for col in df.columns:
if'log'in col.lower() and'fc'in col.lower(): col_map['logfc'] = col
if'adj'in col.lower() or'fdr'in col.lower(): col_map['padj'] = col
if'gene'in col.lower() or'symbol'in col.lower(): col_map['gene'] = col
print(f"检测到的列名映射: {col_map}")
# 3. 设置阈值
pval_threshold = 0.05
logfc_threshold = 1.0
# 4. 数据预处理
# 处理可能的 0 值导致的 infinite log 值
df[col_map['padj']] = df[col_map['padj']].replace(0, 1e-300) # Replace 0 with a very small number
df['nlog10'] = -np.log10(df[col_map['padj']])
# 分组标记
df['Group'] = 'NS'
df.loc[(df[col_map['padj']] < pval_threshold) & (df[col_map['logfc']] > logfc_threshold), 'Group'] = 'Up'
df.loc[(df[col_map['padj']] < pval_threshold) & (df[col_map['logfc']] < -logfc_threshold), 'Group'] = 'Down'
# 5. 绘图设置 (SCI 风格)
plt.figure(figsize=(8, 6), dpi=300)
sns.set_style("ticks")
plt.rcParams['font.family'] = 'sans-serif'
colors = {'NS': '#bdbdbd', 'Up': '#E64B35', 'Down': '#3C5488'}
sns.scatterplot(data=df, x=col_map['logfc'], y='nlog10',
hue='Group', palette=colors,
style='Group', markers={'Up':'o', 'Down':'o', 'NS':'o'},
edgecolor='none', alpha=0.8, s=20, legend='full')
# 6. 添加辅助线
plt.axhline(-np.log10(pval_threshold), color='black', linestyle='--', linewidth=0.8, alpha=0.7)
plt.axvline(logfc_threshold, color='black', linestyle='--', linewidth=0.8, alpha=0.7)
plt.axvline(-logfc_threshold, color='black', linestyle='--', linewidth=0.8, alpha=0.7)
# 7. 坐标轴和标签
plt.title('Volcano Plot', fontsize=14, fontweight='bold')
plt.xlabel(r'$log_{2}(Fold Change)$', fontsize=12)
plt.ylabel(r'$-log_{10}(P_{adj})$', fontsize=12)
sns.despine()
# 8. 标注 Top 基因
if col_map['gene'] in df.columns:
top_genes = pd.concat([
df[df['Group'] == 'Up'].nlargest(5, 'nlog10'),
df[df['Group'] == 'Down'].nlargest(5, 'nlog10')
])
texts = []
for _, row in top_genes.iterrows():
texts.append(plt.text(row[col_map['logfc']], row['nlog10'], row[col_map['gene']], fontsize=8))
try:
from adjustText import adjust_text
adjust_text(texts, arrowprops=dict(arrowstyle='-', color='black', lw=0.5))
except ImportError:
print("提示: 未安装 adjustText 库,基因标签可能会重叠。建议 pip install adjustText")
except Exception as e:
print(f"标注调整时发生错误: {e}")
# 9. 保存图片
plt.tight_layout()
plt.savefig(f'{output_name}.png', dpi=300, bbox_inches='tight')
plt.savefig(f'{output_name}.pdf', dpi=300, bbox_inches='tight')
print(f"绘图完成!已保存为 {output_name}.png 和 {output_name}.pdf")
if __name__ == "__main__":
plot_sci_volcano('05.DE/de_result.tsv')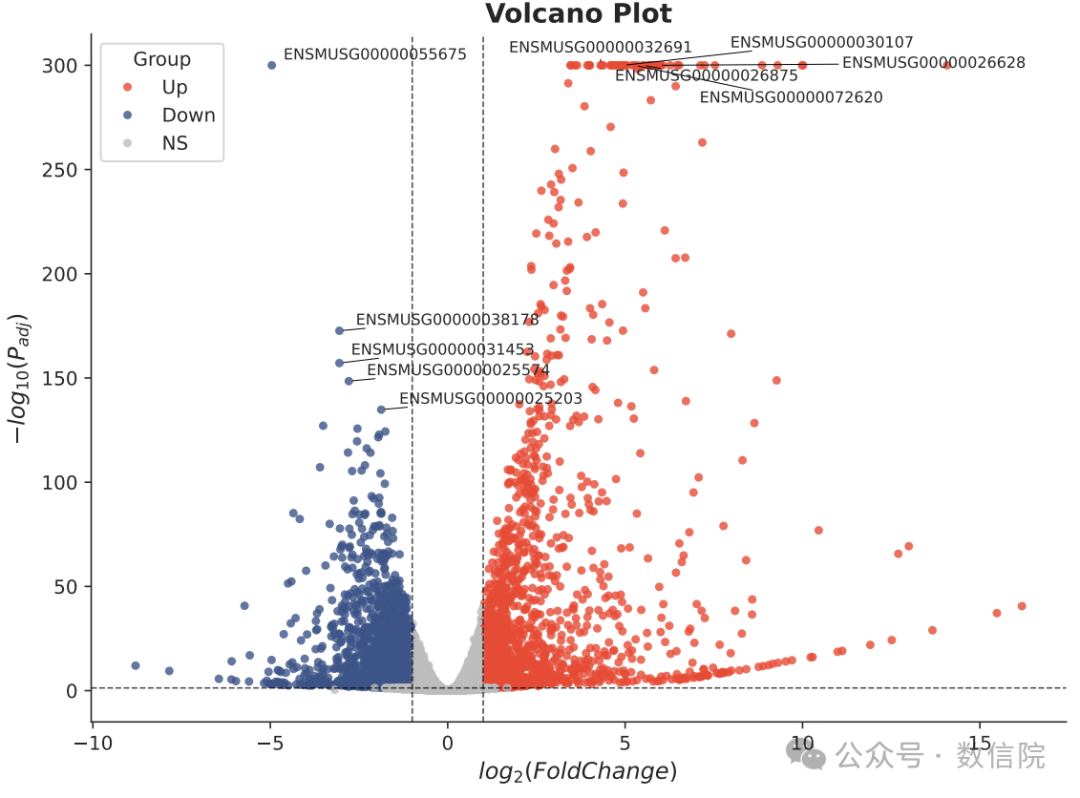
换成R语言绘图
下面是gemini给出的代码和运行的结果
# 加载必要的库
if (!require("ggplot2")) install.packages("ggplot2", repos="http://cran.us.r-project.org")
if (!require("ggrepel")) install.packages("ggrepel", repos="http://cran.us.r-project.org")
if (!require("dplyr")) install.packages("dplyr", repos="http://cran.us.r-project.org")
library(ggplot2)
library(ggrepel)
library(dplyr)
# 1. 读取数据
file_path <- "05.DE/de_result.tsv"
data <- read.table(file_path, header=TRUE, sep="\t", stringsAsFactors=FALSE)
# 2. 设置阈值
pval_threshold <- 0.05
logfc_threshold <- 1.0
# 3. 数据处理
# 填充 NA 和处理 0 值
data$padj[is.na(data$padj)] <- 1
min_nonzero <- min(data$padj[data$padj > 0], na.rm=TRUE)
if (is.infinite(min_nonzero)) min_nonzero <- 1e-300
data$padj[data$padj == 0] <- min_nonzero / 10
data$nlog10 <- -log10(data$padj)
# 分组
data$Group <- "NS"
data$Group[data$padj < pval_threshold & data$log2FoldChange > logfc_threshold] <- "Up"
data$Group[data$padj < pval_threshold & data$log2FoldChange < -logfc_threshold] <- "Down"
data$Group <- factor(data$Group, levels=c("Up", "Down", "NS"))
# 计算上下调基因数量,用于显示
count_up <- sum(data$Group == "Up")
count_down <- sum(data$Group == "Down")
# 4. 筛选 Top 基因 (增加筛选逻辑:结合 logFC 和 pvalue 的重要性)
# 定义一个 "重要性分数" = logFC绝对值 * -log10(pvalue)
data$importance <- abs(data$log2FoldChange) * data$nlog10
top_genes <- data %>%
filter(Group != "NS") %>%
arrange(desc(importance)) %>%
slice_head(n = 10) # 取前 10 个最"重要"的基因
# 5. 高级风格设置 (Nature Publishing Group 风格配色 + 现代排版)
# 手动定义 NPG 风格颜色
color_palette <- c("Up" = "#E64B35", # 朱红
"Down" = "#00A087", # 青绿 (Teal)
"NS" = "#d9d9d9") # 极浅灰,降低干扰
# 动态设置坐标轴范围,保证对称美观
max_x <- max(abs(data$log2FoldChange), na.rm=TRUE)
limit_x <- ceiling(max_x) + 1
p <- ggplot(data, aes(x=log2FoldChange, y=nlog10)) +
# A. 主散点层
# 使用 shape=21 (带边框的圆点),可以让点看起来更精致
# size 映射到显著性 (nlog10),越显著点越大,增加层次感
geom_point(aes(fill=Group, size=nlog10),
color="white", # 点的边框颜色为白色
stroke=0.2, # 边框极细
shape=21, # 填充型圆点
alpha=0.85) + # 轻微透明
# B. 颜色与大小映射设置
scale_fill_manual(values=color_palette) +
scale_size_continuous(range = c(1.5, 4.5), guide = "none") + # 限制点的大小范围,并不显示大小图例
# C. 辅助线 (更优雅的浅灰色)
geom_vline(xintercept=c(-logfc_threshold, logfc_threshold), linetype="longdash", color="#7f7f7f", alpha=0.5, size=0.4) +
geom_hline(yintercept=-log10(pval_threshold), linetype="longdash", color="#7f7f7f", alpha=0.5, size=0.4) +
# D. 统计信息注释 (直接标在图上,不需要看图例数数)
annotate("text", x = -limit_x * 0.8, y = max(data$nlog10) * 0.95,
label = paste0("Down-regulated\nn = ", count_down),
color = color_palette["Down"], fontface = "bold", size = 5, hjust=0) +
annotate("text", x = limit_x * 0.8, y = max(data$nlog10) * 0.95,
label = paste0("Up-regulated\nn = ", count_up),
color = color_palette["Up"], fontface = "bold", size = 5, hjust=1) +
# E. 坐标轴与排版
labs(title="Differential Expression Analysis",
subtitle=paste0("Total genes: ", nrow(data), " | Cutoff: p<0.05, |logFC|>1"),
x=expression(log[2]("Fold Change")),
y=expression(-log[10]("P-adjusted"))) +
xlim(-limit_x, limit_x) + # 强制对称 X 轴
theme_minimal(base_size = 14) + # 使用极简主题为基础
theme(
plot.title = element_text(hjust = 0.5, face = "bold", size = 18),
plot.subtitle = element_text(hjust = 0.5, color = "grey40", size = 10, margin = margin(b = 20)),
panel.grid.major = element_line(color = "grey92", size = 0.5), # 极淡的网格
panel.grid.minor = element_blank(),
axis.line = element_line(color = "black", size = 0.5), # 坐标轴线
axis.text = element_text(color = "black"),
legend.position = "none"# 隐藏图例 (因为图上已经标了 Up/Down 文字)
) +
# F. 基因标签 (使用 geom_label_repel 带背景框,更清晰)
geom_label_repel(data=top_genes, aes(label=gene_id, color=Group),
fill="white", # 标签背景色
fontface="bold",
size=3,
box.padding=0.5,
point.padding=0.5,
min.segment.length=0, # 总是显示连接线
segment.color="grey50",
segment.size=0.5,
max.overlaps=Inf,
show.legend=FALSE) +
scale_color_manual(values=color_palette) # 标签文字颜色跟随点的颜色
# 6. 保存
ggsave("volcano_plot_chic.png", plot=p, width=8, height=7, dpi=300)
ggsave("volcano_plot_chic.pdf", plot=p, width=8, height=7)
print("高级风格绘图完成!已保存为 volcano_plot_chic.png")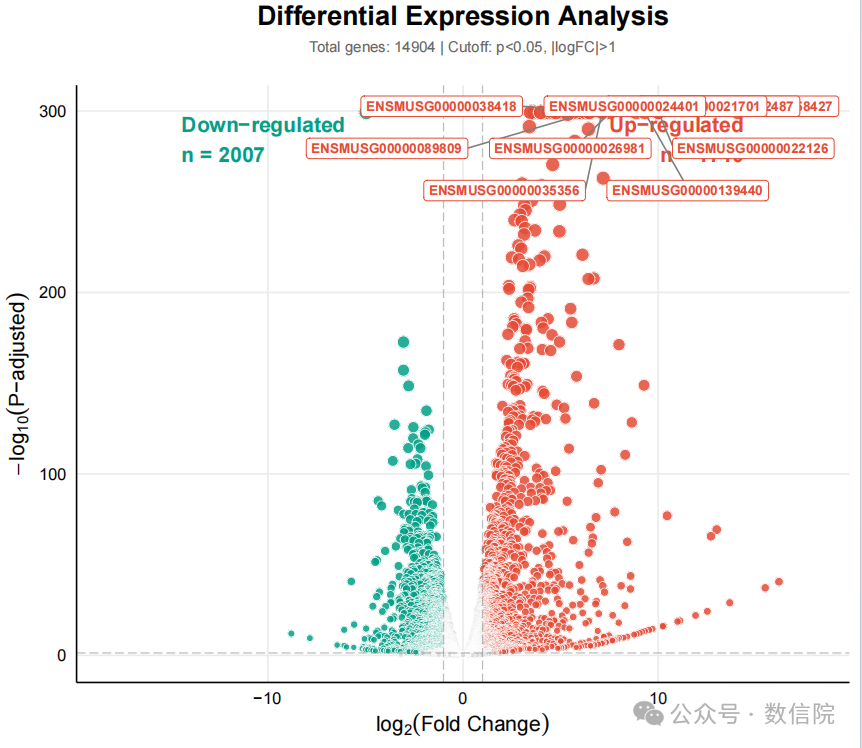
Gemini 3 最适合生成哪些科研图?
物信息学
-
PCA / UMAP / t-SNE
-
火山图、热图
-
ChIP-seq / ATAC-seq peak 分布图
-
RNA-seq 差异基因分析图
统计模型
-
回归曲线
-
随机森林特征重要性
-
ROC / PR 曲线
写在最后
未来的大模型(包括 Gemini 3)会做到:
⭐ 你只需要一句话
生成适用于《Nature》投稿的图,格式按照Nature标准。
大模型会自动:
-
读取原始数据
-
分析
-
绘图
-
标注
-
排版
-
输出 PDF/AI 矢量图
-
生成图注 + 说明
科研绘图不再是瓶颈,而是:
🔥 最高效、最轻松的环节之一。