引言
空间转录组(Spatial Transcriptomics, ST)技术在传统转录组与单细胞转录组的基础上,引入了空间位置信息 ,使研究者能够在组织结构背景下解析基因表达模式。相较于下游分析中对空间结构、生物学机制的深入挖掘,上游分析的核心目标在于:将测序原始数据转换为高质量、可用于空间建模和生物学解释的表达矩阵与空间坐标信息。
空间转录组的上游分析直接决定了后续空间聚类、空间差异基因、空间通讯等分析的可靠性。因此,系统、规范地理解其上游分析流程具有重要意义。
空间转录组数据类型概述
不同空间转录组技术路线,其上游数据形态与处理方式存在一定差异,但总体可归纳为以下几类:
1. 基于捕获芯片的空间转录组(如 10x Visium)
-
原始数据:FASTQ
-
空间信息来源:芯片上固定位置的 barcode
-
特点:一个 spot 通常包含多个细胞
2. 原位杂交/成像型空间转录组(如 MERFISH、seqFISH)
-
原始数据:显微图像
-
空间信息来源:细胞或分子级坐标
-
特点:空间分辨率高,基因数相对受限
3. 组织切片测序型(如 Slide-seq、Stereo-seq)
-
原始数据:FASTQ + bead 坐标文件
-
特点:高分辨率、大规模空间点位
本文重点以尤其是 10x Visium为代表,系统介绍其上游分析流程,需要sratoolkit与Space Ranger
安装
sratoolkit官网安装
bash
下载的为Ubuntu Linux X64
wget -c https://ftp-trace.ncbi.nlm.nih.gov/sra/sdk/3.3.0/sratoolkit.3.3.0-ubuntu64.tar.gz
解压即可
tar -zxvf sratoolkit.3.3.0-ubuntu64.tar.gz
可选择添加到环境变量
export PATH=$PATH:$PWD/sratoolkit.3.3.0-ubuntu64/binSpace Ranger官网安装
bash
下载Space Ranger
wget -O spaceranger-4.0.1.tar.gz "https://cf.10xgenomics.com/releases/spatial-exp/spaceranger-4.0.1.tar.gz?Expires=1766082461&Key-Pair-Id=APKAI7S6A5RYOXBWRPDA&Signature=ehvI5hDYu5uCFb36xyvl-97DQS6mnL506M58Xf5uVC4q33IHmkqx8qx81Ifm3-xAMhKz453qkl~onEejVL~rVQsW4Dtf32sJkDyTBoQtG8WDzTJuoGQk9uIpszGovFixWWuluoxvH2bYAJZH90yNTVz746Iq3DXuveCD0j2gEUiB20~DTPfPkWoMPACy1B2Vd2l2kUE-aG2NcCLtgvvYA0ktj9~SEf299BaD19tkZlWbIvQPbkJomCIG4csLr~69UmTKjOpr~K-1TNw1gc1cq429uREdJJMMlzd6PWfgaIPZu9wLCUJJVcBCcyuYtiJmFUUa79NTInJXei1RbMWPvQ__"
解压
tar -zxvf spaceranger-4.0.1.tar.gz
添加到环境变量
export PATH=$PATH:$PWD/spaceranger-4.0.1-ubuntu64/bin
下载参考基因组
人
wget "https://cf.10xgenomics.com/supp/cell-exp/refdata-gex-GRCh38-2024-A.tar.gz"
解压
tar -zxvf refdata-gex-GRCh38-2024-A.tar.gz
Mouse
wget "https://cf.10xgenomics.com/supp/cell-exp/refdata-gex-GRCm39-2024-A.tar.gz"
Rat
wget "https://cf.10xgenomics.com/supp/cell-exp/refdata-gex-mRatBN7-2-2024-A.tar.gz"下载数据
选择的数据SRR编号为SRR27277620,GEO编号为GSM7980872,组织类型:甲状腺癌,临床分型:ATC
空间转录组的上游分析不仅需要fastq文件,还需要一个HE组织染色图像。
图像一般都在GEO数据库的补充文件中
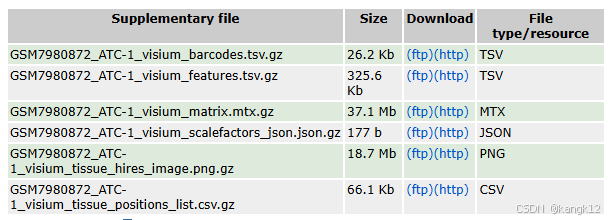
第四个下载解压即可
bash
prefetch --max-size 100G SRR27277620
将下载的sra转为fastq
fasterq-dump SRR27277612.sra -O fastq_store -e 20 --include-technical
修改名字
mv SRR27277612_1.fastq SRR27277612_S1_L001_R1_001.fastq
mv SRR27277612_2.fastq SRR27277612_S1_L001_R2_001.fastq注意sra转为fastq过程中:--include-technical → 保留空间转录组必需的技术读段
转完格式之后,必须将fastq名称修改为Space Ranger规定的格式
bash
<样本名>_<S编号>_<Lane编号>_<Read类型>_<001>.fastq.gz| 组成部分 | 格式示例 | 含义说明 | 必需性 | Spaceranger 匹配规则 |
|---|---|---|---|---|
| 样本名 | PTC, sample1 |
样本标识符,与 --sample 参数一致 |
必需 | 通过 --sample 指定前缀 |
| S编号 | S1, S2 |
样本在测序中的编号 | 通常必需 | 自动识别,无需指定 |
| Lane编号 | L001, L002 |
测序 lane 编号 | 通常必需 | 自动按 lane 分组配对 |
| Read类型 | R1, R2, I1 |
读段类型 | 必需 | 自动配对 R1↔R2 |
| 文件编号 | 001, 002 |
文件批次编号 | 通常为001 | 自动识别 |
一般只有R1和R2两个fastq文件,也有的会有I1文件
| 文件类型 | 标准命名 | 主要功能 | 内容长度 | 在空间转录组中的用途 |
|---|---|---|---|---|
| R1 (Read 1) | *_R1_*.fastq.gz |
空间barcode + UMI | 28 bp (Visium v1) | 识别spot位置和分子计数 |
| R2 (Read 2) | *_R2_*.fastq.gz |
cDNA序列 | 50-150 bp | 基因识别和定量 |
| I1 (Index 1) | *_I1_*.fastq.gz |
样本index | 8-10 bp | 多样本混合时区分样本 |
上游分析
前期处理好之后就开始进行空转上游处理
在运行 spaceranger count 时,组织图像与芯片坐标的对齐(image alignment)是一个关键步骤。Space Ranger 实际上提供了两种对齐策略:
- 自动对齐(Automatic alignment):由 Space Ranger 在 count 过程中自动完成
- 手动对齐(Manual alignment):借助 Loupe Browser 预先完成人工校准,再由 Space Ranger 读取结果
自动对齐
bash
spaceranger count \
--id=ATC \
--transcriptome=/home/duyo/data_251215/huma_data/refdata-gex-GRCh38-2024-A \
--fastqs=/home/duyo/data_251215/SRR27277612data/SRR27277612/fastq_store/ \
--sample=SRR27277612 \
--image=./GSM7980872_ATC-1_visium_tissue_hires_image.png \
--unknown-slide visium-1 \
--localcores=16 \
--localmem=64 \
--create-bam false- **
--id**分析任务名称,同时作为输出目录名。 - **
--transcriptome**指定 10x 官方格式的人类参考转录组(GRCh38)。 - **
--fastqs**FASTQ 文件所在目录。 - **
--sample**指定需要分析的样本名,用于匹配 FASTQ 文件。 - **
--image**组织切片图像(H&E),用于空间对齐和 in-tissue 判定。 - **
--unknown-slide**指定芯片类型为标准 10x Visium(无芯片序列号时使用)。
如果知道芯片编号可以使用 --slide=V19J01-123
不知道芯片序列需要使用**--unknown-slide** 选择芯片的类型**,后面必须指定以下之一**
visium-1→ 第一代 Visium 载玻片(标准 6.5mm 捕获区)visium-2→ 第二代 Visium 载玻片(新版设计)visium-2-large→ 第二代大尺寸载玻片visium-hd→ 高分辨率 Visium HD 载玻片
- **
--localcores**使用多少个 CPU 核心进行计算。 - **
--localmem**分配多少内存供分析使用。 - **
--create-bam false**不生成 BAM 文件,以节省磁盘空间。
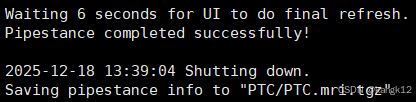
当看到这几行时说明运行成功了!!
手动对齐
需要下载Loupe Browser进行对齐,下载连接与教程连接
Loupe Browser | Official 10x Genomics Support![]() https://www.10xgenomics.com/support/software/loupe-browser/latest对齐之后会输出一个json文件,使用--loupe-alignment指定该文件
https://www.10xgenomics.com/support/software/loupe-browser/latest对齐之后会输出一个json文件,使用--loupe-alignment指定该文件
例如
bash
spaceranger count \
--id=ATC \
--transcriptome=/home/duyo/data_251215/huma_data/refdata-gex-GRCh38-2024-A \
--fastqs=/home/duyo/data_251215/SRR27277612data/SRR27277612/fastq_store/ \
--sample=SRR27277612 \
--image=./GSM7980872_ATC-1_visium_tissue_hires_image.png \
--unknown-slide visium-1 \
--localcores=16 \
--localmem=64 \
--create-bam false \
-loupe-alignment=SRR27277612.json输出内容说明
不同的芯片格式输出内容不同(所以分析时最好有芯片序列号),参考官网说明
使用芯片Visium HD 或 Visium HD'3
| 文件或目录名称 | 描述 |
|---|---|
barcode_mappings.parquet |
该文件高效存储空间映射信息,本质上作为CSV文件,追踪Visium HD数据中条码(方块)、核、单元和箱之间的关系。详情请参见分段输出页面。 |
binned_outputs |
默认情况下,该目录有三个子目录:、、和。每个目录包含 、 、 、 和 。该目录仅提供8微米和16微米的频箱尺寸。仅提供8微米的箱体尺寸。仅提供2微米分辨率。square_002um``square_008um``square_016um``filtered_feature_bc_matrix``raw_feature_bc_matrix``spatial``filtered_feature_bc_matrix.h5``raw_feature_bc_matrix.h5``analysis``cloupe.cloupe``raw_probe_bc_matrix.h5 |
cloupe_008um.cloupe |
与 .cloupe 文件的 8 微米 bin 尺寸有对称链接 |
cloupe_cell.cloupe |
与.cloupe文件的单元格分段的对称链接 |
feature_slice.h5 |
一种专为 Visium HD 设计的新文件类型,支持高效获取单个或多个基因的 2 微米分辨率图像切片。详情请见此页面。 |
metrics_summary.csv |
以CSV格式运行汇总指标 |
molecule_info.h5 |
包含所有含有有效条形码、有效UMI且高度确定分配给基因条码或bin的分子的每分子信息。 |
probe_set.csv |
输入探针集的副本,参考CSV文件。 |
segmented_outputs |
包含分段输出的文件夹。包含 , , , , , 和 。详情请参见分段输出页面。analysis``cell_segmentations.geojson``cloupe.cloupe``filtered_feature_cell_matrix``filtered_feature_cell_matrix.h5``graphclust_annotated_cell_segmentations.geojson``graphclust_annotated_nucleus_segmentations.geojson``nucleus_segmentations.geojson``raw_feature_cell_matrix``raw_feature_cell_matrix.h5``spatial |
spatial |
包含数据空间性的输出文件夹。更多详情请参见空间输出页面。 |
web_summary.html |
以HTML格式运行汇总指标和图表 |
使用Visium v1/v2
| 文件或目录名称 | 描述 |
|---|---|
web_summary.html |
以HTML格式运行汇总指标和图表 |
cloupe.cloupe |
放大镜浏览器可视化与分析文件 |
spatial/ |
包含数据空间性的输出文件夹。 |
analysis/ |
包含次级分析数据的文件夹,包括基于图的聚类和K均值聚类(K = 2-10);簇间的基因表达差异;PCA、t-SNE和UMAP降维。 |
metrics_summary.csv |
以CSV格式运行汇总指标 |
probe_set.csv |
输入探针集的副本,参考CSV文件。关于Visium FFPE和CytAssist工作流程的呈现 |
possorted_genome_bam.bam |
索引BAM文件,包含位置排序的读段,与基因组和转录组对齐,并附有条形码信息 |
possorted_genome_bam.bam.bai |
索引。如果参考转录组是从染色体非常长的基因组(>512 Mbp)生成的,Space Ranger v2.0+ 会生成索引文件。possorted_genome_bam.bam``possorted_genome_bam.bam.csi |
filtered_feature_bc_matrix/ |
仅包含MEX格式的组织相关条码。矩阵中的每个元素分别是与特征(行)和条码(列)相关的UMI数量。该文件可以输入第三方软件包,允许用户作条码特征矩阵(例如过滤异常点、运行降维、规范基因表达)。 |
filtered_feature_bc_matrix.h5 |
信息与HDF5格式相同。filtered_feature_bc_matrix/ |
raw_feature_bc_matrices/ |
包含所有检测到的MEX格式条码。矩阵中的每个元素分别是与特征(行)和条码(列)相关的UMI数量。 |
raw_feature_bc_matrix.h5 |
信息与HDF5格式相同。raw_feature_bc_matrices/ |
raw_probe_bc_matrix.h5 |
包含所有检测到的条码的每个探头的UMI计数,格式为HDF5格式。仅在运行探针检测管道时生产。 |
molecule_info.h5 |
包含所有含有有效条形码、有效UMI且高度置信度地分配给基因或蛋白质条码的分子的每分子信息。该文件对于包括 、 和 在内的其他分析管道是必需的。spaceranger``aggr``targeted-compare``targeted-depth |
但是一般分析完成之后,我们所需的下游分析所需文件主要集中于outs文件
bash
outs
├── aggregation.csv
├── aggr_tissue_positions.csv
├── analysis
│ ├── clustering
│ ├── diffexp
│ ├── pca
│ ├── tsne
│ └── umap
├── cloupe.cloupe
├── filtered_feature_bc_matrix
│ ├── barcodes.tsv.gz
│ ├── features.tsv.gz
│ └── matrix.mtx.gz
├── filtered_feature_bc_matrix.h5
├── spatial
│ ├── LV123
│ │ ├── scalefactors_json.json
│ │ ├── tissue_hires_image.png
│ │ └── tissue_lowres_image.png
│ ├── LB456
│ │ ├── scalefactors_json.json
│ │ ├── tissue_hires_image.png
│ │ └── tissue_lowres_image.png
│ └── LP789
│ ├── scalefactors_json.json
│ ├── tissue_hires_image.png
│ └── tissue_lowres_image.png
├── summary.json
└── web_summary.html