字节跳动 PXDesign:AI 设计蛋白,82% 命中率惊艳业界
目录
- MORE 框架通过同时从原子、官能团、分子整体和 3D 结构四个层面学习,构建了更全面的分子表示,为 AI 药物发现提供了强大的新基础模型。
- PXDesign 整合了两种互补的生成模型和一套创新的多预测器筛选策略,大幅提升了从头设计蛋白结合物的实验成功率,并且已经开源。
- ConforFormer 通过对比学习,从分子的多种 3D 构象中提炼出一种通用的、与构象无关的向量表示,显著提升了分子属性预测的准确性。
- 通过在史无前例的 3.5 亿细胞数据集上预训练,scPRINT-2 为单细胞分析设定了新的性能天花板,尤其在跨物种泛化能力上表现突出。
- GoMS 通过构建官能团层级的图网络,精准捕捉它们的空间排布和相互作用,解决了传统图神经网络在大分子属性预测上的短板。
!外链图片转存失败,源站可能有防盗链机制,建议将图片保存下来直接上传(https://img-home.csdnimg.cn/images/20230724024159.png?origin_url=data%3Aimage%2Fsvg%2Bxml%2C%253C%253Fxml version%3D'1.0' encoding%3D'UTF-8'%253F%253E%253Csvg width%3D'1px' height%3D'1px' viewBox%3D'0 0 1 1' version%3D'1.1' xmlns%3D'http%3A%2F%2Fwww.w3.org%2F2000%2Fsvg' xmlns%3Axlink%3D'http%3A%2F%2Fwww.w3.org%2F1999%2Fxlink'%253E%253Ctitle%253E%253C%2Ftitle%253E%253Cg stroke%3D'none' stroke-width%3D'1' fill%3D'none' fill-rule%3D'evenodd' fill-opacity%3D'0'%253E%253Cg transform%3D'translate(-249.000000%2C -126.000000\&pos_id=img-Ry5zAUzp-1766808637395)' fill='%23FFFFFF'%3E%3Crect x='249' y='126' width='1' height='1'%3E%3C/rect%3E%3C/g%3E%3C/g%3E%3C/svg%3E)
1. AI 制药新范式:多层次分子预训练模型 MORE
!外链图片转存失败,源站可能有防盗链机制,建议将图片保存下来直接上传(https://img-home.csdnimg.cn/images/20230724024159.png?origin_url=data%3Aimage%2Fsvg%2Bxml%2C%253C%253Fxml version%3D'1.0' encoding%3D'UTF-8'%253F%253E%253Csvg width%3D'1px' height%3D'1px' viewBox%3D'0 0 1 1' version%3D'1.1' xmlns%3D'http%3A%2F%2Fwww.w3.org%2F2000%2Fsvg' xmlns%3Axlink%3D'http%3A%2F%2Fwww.w3.org%2F1999%2Fxlink'%253E%253Ctitle%253E%253C%2Ftitle%253E%253Cg stroke%3D'none' stroke-width%3D'1' fill%3D'none' fill-rule%3D'evenodd' fill-opacity%3D'0'%253E%253Cg transform%3D'translate(-249.000000%2C -126.000000\&pos_id=img-7DuiCT3k-1766808637395)' fill='%23FFFFFF'%3E%3Crect x='249' y='126' width='1' height='1'%3E%3C/rect%3E%3C/g%3E%3C/g%3E%3C/svg%3E)
在 AI 制药领域,我们总是在寻找更好的方式来让机器「读懂」分子。一个好的分子表示 (molecular representation) 是所有下游任务的基石,从预测活性到毒性,无一例外。
过去很多模型只盯着分子的一部分看。有的看 2D 图谱,就像看一张平面设计图;有的只关注 3D 构象。这就像盲人摸象,每个人都只摸到了一部分,得到的理解自然是片面的。
这篇来自 AAAI-25 的论文提出的 MORE 框架,思路就很直接:为什么不把所有信息都用上呢?
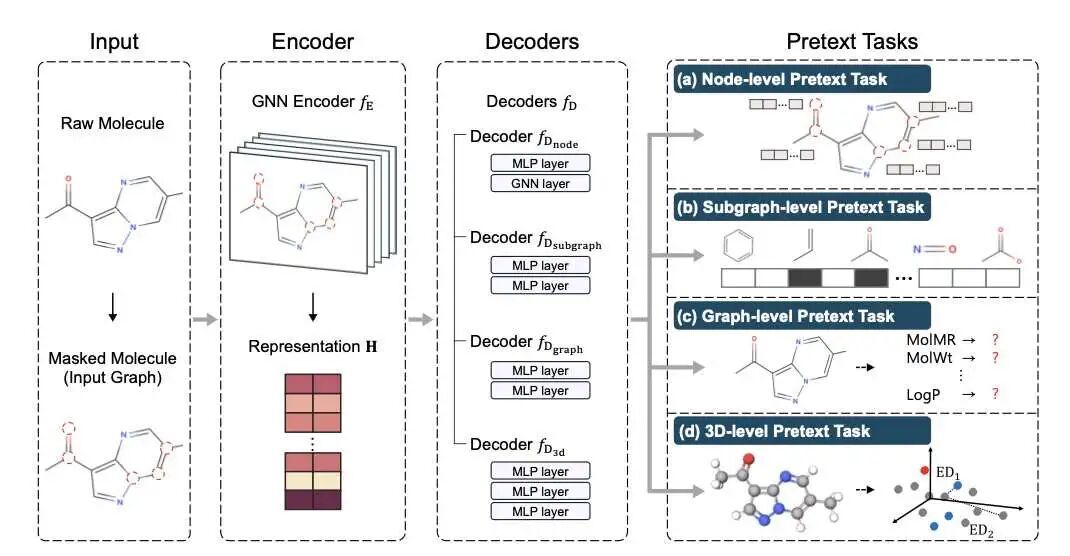
MORE 就像一个经验丰富的化学家,它会从四个层面同时审视一个分子。
- 原子层面 (Node-level): 它会看每个原子的基本属性,这是最基础的化学文法。
- 子图层面 (Subgraph-level): 然后它会识别出像苯环、羧基这样的关键功能团 (functional groups)。这些是决定分子「性格」的关键模块。
- 分子图层面 (Graph-level): 接着,它会把分子作为一个整体来看,理解原子和功能团是如何连接成一个网络的。
- 3D 结构层面 (3D-level): 最后,它还会考虑分子的三维空间构象。毕竟,药物和靶点的结合是在三维空间里发生的,形状至关重要。
为了实现这一点,研究者设计了一个编码器 - 解码器 (encoder-decoder) 架构,用一个编码器学习统一的表示,然后用四个专门的解码器分别处理这四个层面的任务。
我个人认为,MORE 最亮眼的一点是它引入了「分子描述符」 (molecular descriptors) 这类高级语义信息。以前的预训练模型基本都忽略了这一点。分子描述符是什么?它们是化学家们总结出来的经验规律,比如分子量、脂水分配系数 (LogP)、极性表面积 (PSA) 等。这些是我们在药物研发中天天都要看的参数,直接关系到分子的成药性 (druggability)。把这些「化学家直觉」教给模型,效果立竿见影,尤其是在预测毒性和结合亲和力这类复杂任务上。
数据显示,无论是在线性探测 (linear probing) 还是完全微调 (full fine-tuning) 的场景下,MORE 在多个下游任务上的表现都超过了现有模型。
还有一个有趣的发现:在微调过程中,MORE 的参数变化很小。这意味着预训练阶段学到的知识非常扎实,没有在针对新任务微调时被「冲刷」掉。这对于构建一个真正通用的基础模型来说,是至关重要的特性。
研究者还做了扩展性实验,发现随着预训练数据集的增大,MORE 的性能还能持续提升。这给我们描绘了一个美好的前景:如果用海量分子数据来训练一个超大规模的 MORE 模型,它有没有可能成为化学领域的 GPT?一个能回答关于分子各种问题的基础模型?
📜Title: More: Molecule Pretraining with Multi-Level Pretext Task
🌐Paper: https://ojs.aaai.org/index.php/AAAI/article/download/34262/36417
!外链图片转存失败,源站可能有防盗链机制,建议将图片保存下来直接上传(https://img-home.csdnimg.cn/images/20230724024159.png?origin_url=data%3Aimage%2Fsvg%2Bxml%2C%253C%253Fxml version%3D'1.0' encoding%3D'UTF-8'%253F%253E%253Csvg width%3D'1px' height%3D'1px' viewBox%3D'0 0 1 1' version%3D'1.1' xmlns%3D'http%3A%2F%2Fwww.w3.org%2F2000%2Fsvg' xmlns%3Axlink%3D'http%3A%2F%2Fwww.w3.org%2F1999%2Fxlink'%253E%253Ctitle%253E%253C%2Ftitle%253E%253Cg stroke%3D'none' stroke-width%3D'1' fill%3D'none' fill-rule%3D'evenodd' fill-opacity%3D'0'%253E%253Cg transform%3D'translate(-249.000000%2C -126.000000\&pos_id=img-YFE8OQVW-1766808637396)' fill='%23FFFFFF'%3E%3Crect x='249' y='126' width='1' height='1'%3E%3C/rect%3E%3C/g%3E%3C/g%3E%3C/svg%3E)
2. 字节跳动 PXDesign:AI 设计蛋白,82% 命中率惊艳业界
!外链图片转存失败,源站可能有防盗链机制,建议将图片保存下来直接上传(https://img-home.csdnimg.cn/images/20230724024159.png?origin_url=data%3Aimage%2Fsvg%2Bxml%2C%253C%253Fxml version%3D'1.0' encoding%3D'UTF-8'%253F%253E%253Csvg width%3D'1px' height%3D'1px' viewBox%3D'0 0 1 1' version%3D'1.1' xmlns%3D'http%3A%2F%2Fwww.w3.org%2F2000%2Fsvg' xmlns%3Axlink%3D'http%3A%2F%2Fwww.w3.org%2F1999%2Fxlink'%253E%253Ctitle%253E%253C%2Ftitle%253E%253Cg stroke%3D'none' stroke-width%3D'1' fill%3D'none' fill-rule%3D'evenodd' fill-opacity%3D'0'%253E%253Cg transform%3D'translate(-249.000000%2C -126.000000\&pos_id=img-ljDYot5c-1766808637396)' fill='%23FFFFFF'%3E%3Crect x='249' y='126' width='1' height='1'%3E%3C/rect%3E%3C/g%3E%3C/g%3E%3C/svg%3E)
在药物研发领域,从头设计一个能精确结合靶点的蛋白质,就像是为一个只见过照片的锁,凭空打造一把钥匙。许多计算方法在电脑里看起来很完美,但一到实验室里就现了原形。这就是为什么看到 PXDesign 的实验数据时,会让人眼前一亮。
两种生成策略,覆盖不同需求
研究者们没有把宝押在一种算法上,而是用了两条腿走路。
第一条路是 PXDesign-d,它基于扩散模型 (diffusion model)。你可以把它想象成一位极富创造力的雕塑家。它从一团随机的「数字黏土」开始,一步步把它雕琢成一个合理的蛋白质骨架。这种方法的优点在于能生成结构非常新颖、多样性极高的分子,有可能发现自然界中从未出现过的全新骨架。这对探索未知的设计空间很有用。
第二条路是 PXDesign-h,基于幻觉模型 (hallucination model)。这个更像一个目标明确的工程师。你告诉它:「我需要在蛋白质的这个位置放上一个能结合靶点的特定结构(motif)。」然后,模型就会围绕这个核心功能区,构建出一个稳定可折叠的蛋白质框架。这种方法非常适合针对一个已知的结合位点进行精细优化。
真正的亮点:用多个「裁判」来筛选
生成成千上万个候选分子在今天已经不算难事,真正的瓶颈在于如何从沙子里淘出金子。绝大多数设计的分子在现实中要么无法正确折叠,要么根本没有活性。
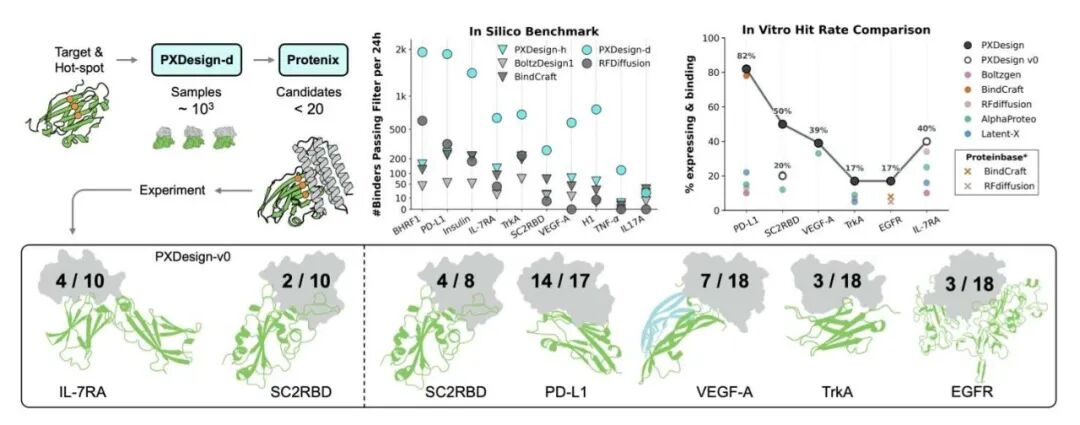
PXDesign 在这里的做法非常聪明。他们没有只依赖 AlphaFold 这一个「裁判」来判断设计的结构好不好。相反,他们组建了一个「裁判团」,里面包括了自研的 Protenix 和多个不同版本的 AlphaFold 模型。
这背后的逻辑很简单。每个结构预测模型都有自己的偏好和盲点。一个设计如果只在一个模型上得分高,可能是侥幸。但如果它在多个独立的模型那里都获得了高分,那么它在现实世界中能够正确折叠并发挥功能的概率就大大增加了。这就像是做重大决策前,听取了多位不同背景专家的意见。这个系统性的多预测器筛选策略,是他们能取得如此高实验命中率的关键。
惊人的实验结果
最终,数据是检验真理的唯一标准。他们在 7 个有真实生物学意义的靶点上进行了实验验证,包括像 PD-L1 和 VEGF-A 这样的明星靶点。
结果是,7 个靶点里有 6 个都成功找到了纳摩尔 (nanomolar) 亲和力的结合物。命中率从 17% 到 82% 不等。在蛋白质设计领域,17% 的命中率已经算得上是巨大的成功,而 82% 几乎是闻所未闻的。这意味着研发团队可以大大节省在分子合成与湿实验验证上的时间和金钱,把资源集中在最有希望的候选分子上。
超越传统蛋白
这个框架的潜力还不止于此。研究者们还尝试用它来设计环肽 (cyclic peptide) 结合物,并取得了不错的结果。环肽是介于小分子和抗体之间的一类药物,成药潜力巨大。PXDesign 的通用性表明,它未来可能成为一个更广泛的药物分子设计平台。
他们将整个管线和代码都开源了,还提供了网页服务器。
📜Title: PXDesign: Fast, Modular, and Accurate De Novo Design of Protein Binders
🌐Paper: https://www.biorxiv.org/content/10.1101/2025.08.15.670450v3
💻Code: https://github.com/bytedance/PXDesign
💻Server: https://protenix-server.com
!外链图片转存失败,源站可能有防盗链机制,建议将图片保存下来直接上传(https://img-home.csdnimg.cn/images/20230724024159.png?origin_url=data%3Aimage%2Fsvg%2Bxml%2C%253C%253Fxml version%3D'1.0' encoding%3D'UTF-8'%253F%253E%253Csvg width%3D'1px' height%3D'1px' viewBox%3D'0 0 1 1' version%3D'1.1' xmlns%3D'http%3A%2F%2Fwww.w3.org%2F2000%2Fsvg' xmlns%3Axlink%3D'http%3A%2F%2Fwww.w3.org%2F1999%2Fxlink'%253E%253Ctitle%253E%253C%2Ftitle%253E%253Cg stroke%3D'none' stroke-width%3D'1' fill%3D'none' fill-rule%3D'evenodd' fill-opacity%3D'0'%253E%253Cg transform%3D'translate(-249.000000%2C -126.000000\&pos_id=img-OcEUsn3V-1766808637396)' fill='%23FFFFFF'%3E%3Crect x='249' y='126' width='1' height='1'%3E%3C/rect%3E%3C/g%3E%3C/g%3E%3C/svg%3E)
3. ConforFormer: AI 如何从分子 3D 柔性中学习本质
!外链图片转存失败,源站可能有防盗链机制,建议将图片保存下来直接上传(https://img-home.csdnimg.cn/images/20230724024159.png?origin_url=data%3Aimage%2Fsvg%2Bxml%2C%253C%253Fxml version%3D'1.0' encoding%3D'UTF-8'%253F%253E%253Csvg width%3D'1px' height%3D'1px' viewBox%3D'0 0 1 1' version%3D'1.1' xmlns%3D'http%3A%2F%2Fwww.w3.org%2F2000%2Fsvg' xmlns%3Axlink%3D'http%3A%2F%2Fwww.w3.org%2F1999%2Fxlink'%253E%253Ctitle%253E%253C%2Ftitle%253E%253Cg stroke%3D'none' stroke-width%3D'1' fill%3D'none' fill-rule%3D'evenodd' fill-opacity%3D'0'%253E%253Cg transform%3D'translate(-249.000000%2C -126.000000\&pos_id=img-O1N8iTpi-1766808637396)' fill='%23FFFFFF'%3E%3Crect x='249' y='126' width='1' height='1'%3E%3C/rect%3E%3C/g%3E%3C/g%3E%3C/svg%3E)
在药物发现中,我们总是在跟分子的三维结构打交道。一个分子不是一张静态的 2D 图纸,它在溶液中会不断扭转、弯曲,像一个灵活的体操运动员,拥有很多种姿态,我们称之为「构象」 (Conformation)。哪种构象才是与靶点结合的活性构象?这个问题一直很棘手。很多 AI 模型为了简化问题,干脆只看 2D 结构,但这丢失了最关键的 3D 信息。
ConforFormer 这项工作,就是想正面解决这个问题。
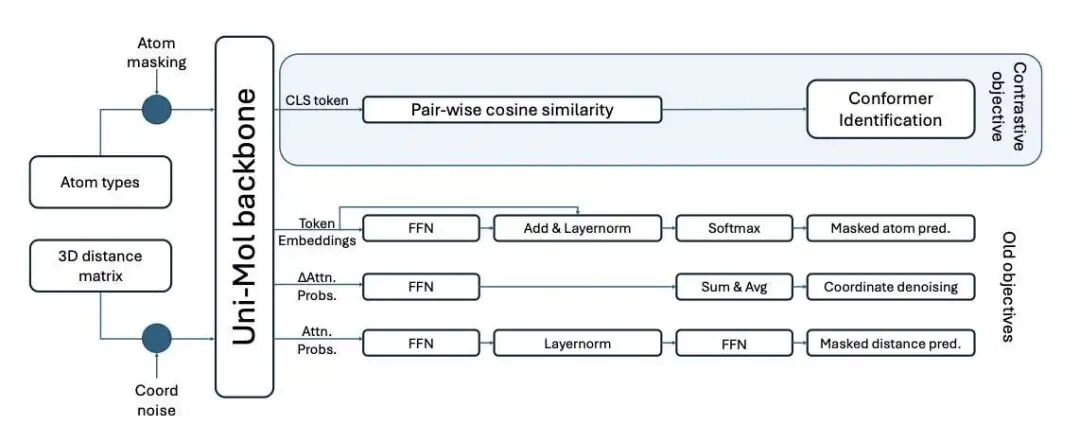
它的核心思路很直观。想象一下,给你看同一个人的多张照片------一张站着,一张坐着,一张在跑步。你一眼就能认出这都是同一个人。ConforFormer 的工作原理也类似。研究者首先为每个分子生成一组合理的、能量较低的 3D 构象。然后,他们用一种叫做对比学习 (contrastive learning) 的方法来训练模型。
这个训练任务很简单:让模型学会把同一个分子的所有不同构象的表示向量(可以理解为机器眼里的「指纹」)拉得尽可能近,同时把不同分子的表示向量推得尽可能远。经过这样训练,模型就学会了忽略构象的细微差异,抓住这个分子的「本质身份」。最终,它能为每个分子生成一个唯一的、与构象无关的表示向量。
这个方法的好处是巨大的。你只需要预训练一次模型,就可以得到一个适用于各种任务的分子表示。无论是预测溶解度、毒性,还是做结构相似性搜索,都可以直接用这个现成的向量,省去了为每个任务或每个构象反复计算的麻烦。
这个模型建立在之前的 Uni-Mol 架构之上,但它的学习目标是全新的。它在训练时甚至不需要看分子的 2D 连接图,只通过学习 3D 几何坐标,就能反推出原子是怎么连接的。这一点非常了不起。研究者发现,ConforFormer 不仅能区分不同构象,还能区分同分异构体 (isomers)------那些化学式相同但结构完全不同的分子。这证明模型学到了类似化学家直觉的东西。
为了验证模型的能力,研究者还专门创建了一个新的基准数据集 PharmIsomer。ConforFormer 在这个数据集上表现出色,说明它在处理药物化学中常见的构象和异构体问题上,确实有两把刷子。对于我们做新药研发的人来说,这意味着我们或许能拥有一个更准确的工具,来做虚拟筛选和分子属性预测,尤其是在那些分子柔性起决定性作用的靶点上。
📜Title: ConforFormer: Representation for Molecules through Understanding of Conformers
🌐Paper: https://doi.org/10.26434/chemrxiv-2025-x68vd
!外链图片转存失败,源站可能有防盗链机制,建议将图片保存下来直接上传(https://img-home.csdnimg.cn/images/20230724024159.png?origin_url=data%3Aimage%2Fsvg%2Bxml%2C%253C%253Fxml version%3D'1.0' encoding%3D'UTF-8'%253F%253E%253Csvg width%3D'1px' height%3D'1px' viewBox%3D'0 0 1 1' version%3D'1.1' xmlns%3D'http%3A%2F%2Fwww.w3.org%2F2000%2Fsvg' xmlns%3Axlink%3D'http%3A%2F%2Fwww.w3.org%2F1999%2Fxlink'%253E%253Ctitle%253E%253C%2Ftitle%253E%253Cg stroke%3D'none' stroke-width%3D'1' fill%3D'none' fill-rule%3D'evenodd' fill-opacity%3D'0'%253E%253Cg transform%3D'translate(-249.000000%2C -126.000000\&pos_id=img-cTkUOtUZ-1766808637396)' fill='%23FFFFFF'%3E%3Crect x='249' y='126' width='1' height='1'%3E%3C/rect%3E%3C/g%3E%3C/g%3E%3C/svg%3E)
4. scPRINT-2: 3.5 亿细胞铸就的细胞基础模型新标杆
!外链图片转存失败,源站可能有防盗链机制,建议将图片保存下来直接上传(https://img-home.csdnimg.cn/images/20230724024159.png?origin_url=data%3Aimage%2Fsvg%2Bxml%2C%253C%253Fxml version%3D'1.0' encoding%3D'UTF-8'%253F%253E%253Csvg width%3D'1px' height%3D'1px' viewBox%3D'0 0 1 1' version%3D'1.1' xmlns%3D'http%3A%2F%2Fwww.w3.org%2F2000%2Fsvg' xmlns%3Axlink%3D'http%3A%2F%2Fwww.w3.org%2F1999%2Fxlink'%253E%253Ctitle%253E%253C%2Ftitle%253E%253Cg stroke%3D'none' stroke-width%3D'1' fill%3D'none' fill-rule%3D'evenodd' fill-opacity%3D'0'%253E%253Cg transform%3D'translate(-249.000000%2C -126.000000\&pos_id=img-9hLhbger-1766808637396)' fill='%23FFFFFF'%3E%3Crect x='249' y='126' width='1' height='1'%3E%3C/rect%3E%3C/g%3E%3C/g%3E%3C/svg%3E)
做单细胞(Single-cell)分析,我们最头疼的是什么?数据噪音大、维度高、还特别稀疏。就像想从一堆混杂着沙子的像素点里,拼凑出一幅清晰的画。这些年,大家都在尝试用大模型来解决这个问题,但总觉得差点意思。
这次,数据量真的成了「护城河」
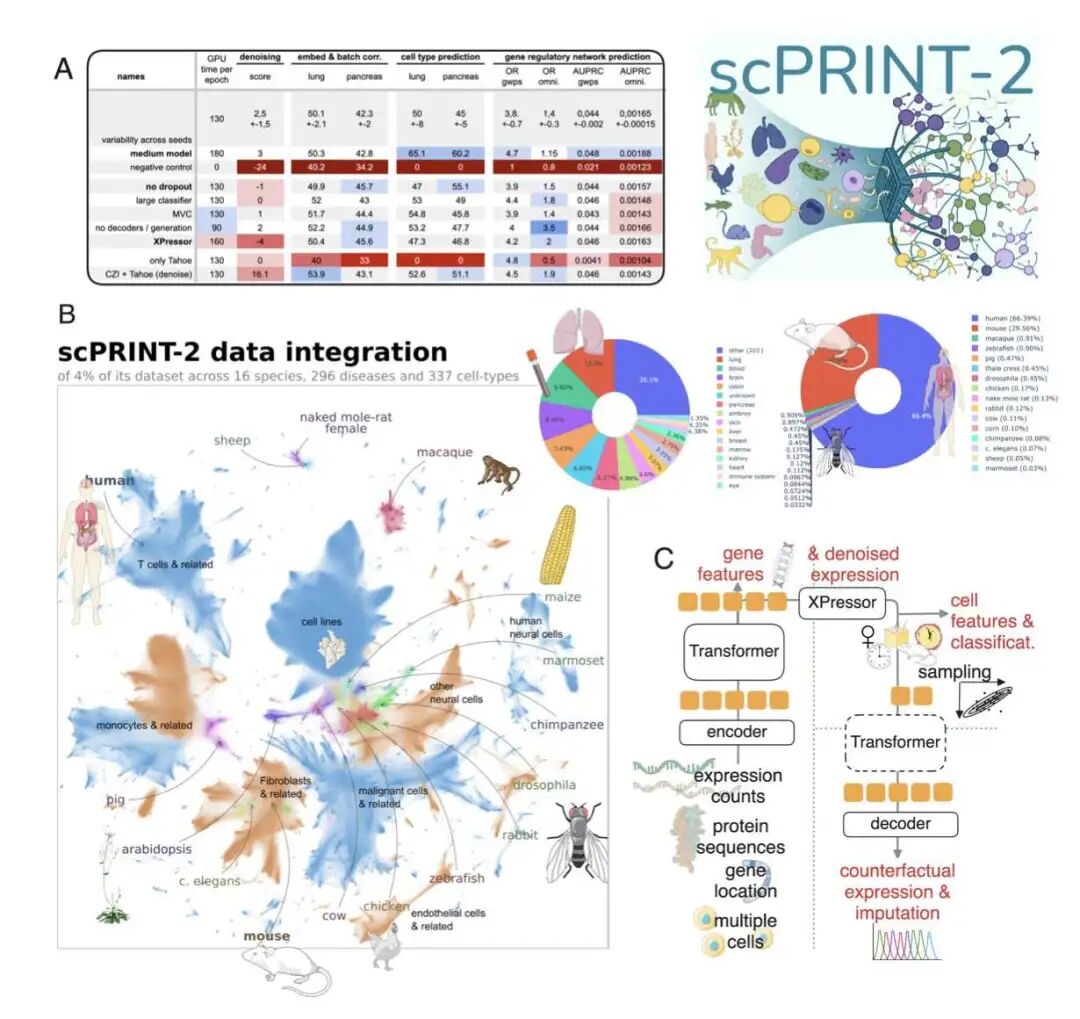
构建一个强大的基础模型,逻辑很简单:给它足够多、足够好的数据。scPRINT-2 的团队就是这么做的,但他们做得非常彻底。他们整合了一个包含 3.5 亿个细胞、横跨 16 个物种的 scRNA-seq(单细胞 RNA 测序)数据集。
这是什么概念?以前的模型可能是在几百万、顶多上千万细胞的数据集上训练的。这就像教一个孩子语言,你是只给他看几本书,还是让他泡在整个互联网里。scPRINT-2 接受的是「互联网级别」的细胞生物学教育。这种规模的数据多样性,让模型有机会学习到超越单个物种或组织的、更底层的生物学规律。这正是实现强大泛化能力的基础。
模型架构:专为单细胞数据量身打造
光有数据还不够,处理工具也得跟上。scPRINT-2 的架构设计有几个特别地方,一看就是懂行的人做的。
首先,它用了一个图神经网络(Graph Neural Network, GNN)编码器。我们都知道,基因不是孤立工作的,它们之间存在复杂的调控网络。传统的模型通常把基因看成一个简单的列表,忽略了这些内在联系。GNN 能够直接对基因间的相互作用关系进行建模,这让模型从一开始就站在了更接近生物学本质的视角上。
其次,是那个新颖的损失函数。单细胞数据里有大量的「零」,很多时候不是因为这个基因不表达,而是因为技术限制没测到,这就是所谓的「dropout」现象。如果用传统的均方误差(Mean Squared Error)来训练,模型会被这些「假零」带偏。scPRINT-2 的研究者把零膨胀负二项(Zero-inflated Negative Binomial)损失和均方误差结合起来。前者是专门处理这种有大量零值的计数数据的统计模型。这相当于给模型配了一副特殊的眼镜,能分清哪些「零」是真没有,哪些「零」是没看到。
真正的考验:在未知领域表现如何?
一个模型好不好,不能只看它在「模拟考」里的分数,得看它在「高考」里的表现。对单细胞模型来说,「高考」就是把它扔到它从未见过的物种或细胞类型中去。
scPRINT-2 在这方面交出的答卷很惊人。它能在没见过的生物(比如涡虫)中准确预测细胞类型和基因网络。这意味着模型学到的不是「小鼠 T 细胞长这样」,而是「什么是 T 细胞」这个更本质的概念。这种跨物种的泛化能力,对于做药物研发的来说价值巨大。我们经常需要在小鼠、猴子等模型动物身上做实验,最终目的是为了应用到人身上。一个能理解物种间共性的模型,就是我们进行跨物种研究的桥梁。
不只是开源代码,更是开放科学
他们不只是把模型权重放出来就完事了。他们把预训练的数据集、任务、甚至训练日志全都公开了。
这为什么重要?因为这让整个研究过程变得透明、可复现。其他人可以清楚地知道这个模型是怎么来的,它的能力边界在哪里,甚至可以在他们的基础上继续优化。这避免了「炼丹黑箱」,为整个领域树立了一个新的标准。当大家都愿意分享自己的「菜谱」时,整个社区的「厨艺」水平才会提高得更快。
scPRINT-2 不是一次小修小补的改进。它通过超大规模的数据、精巧的模型设计和彻底的开放精神,为单细胞基础模型设定了一个新的起点。对于试图在细胞层面理解疾病、寻找新靶点的人来说,这是一个非常强大的新工具。
📜Title: ScPRINT-2: Towards the Next-Generation of Cell Foundation Models and Benchmarks
🌐Paper: https://www.biorxiv.org/content/10.64898/2025.12.11.693702v1
!外链图片转存失败,源站可能有防盗链机制,建议将图片保存下来直接上传(https://img-home.csdnimg.cn/images/20230724024159.png?origin_url=data%3Aimage%2Fsvg%2Bxml%2C%253C%253Fxml version%3D'1.0' encoding%3D'UTF-8'%253F%253E%253Csvg width%3D'1px' height%3D'1px' viewBox%3D'0 0 1 1' version%3D'1.1' xmlns%3D'http%3A%2F%2Fwww.w3.org%2F2000%2Fsvg' xmlns%3Axlink%3D'http%3A%2F%2Fwww.w3.org%2F1999%2Fxlink'%253E%253Ctitle%253E%253C%2Ftitle%253E%253Cg stroke%3D'none' stroke-width%3D'1' fill%3D'none' fill-rule%3D'evenodd' fill-opacity%3D'0'%253E%253Cg transform%3D'translate(-249.000000%2C -126.000000\&pos_id=img-P7hwUJCu-1766808637397)' fill='%23FFFFFF'%3E%3Crect x='249' y='126' width='1' height='1'%3E%3C/rect%3E%3C/g%3E%3C/g%3E%3C/svg%3E)
5. GoMS 架构:看懂分子官能团如何排兵布阵
!外链图片转存失败,源站可能有防盗链机制,建议将图片保存下来直接上传(https://img-home.csdnimg.cn/images/20230724024159.png?origin_url=data%3Aimage%2Fsvg%2Bxml%2C%253C%253Fxml version%3D'1.0' encoding%3D'UTF-8'%253F%253E%253Csvg width%3D'1px' height%3D'1px' viewBox%3D'0 0 1 1' version%3D'1.1' xmlns%3D'http%3A%2F%2Fwww.w3.org%2F2000%2Fsvg' xmlns%3Axlink%3D'http%3A%2F%2Fwww.w3.org%2F1999%2Fxlink'%253E%253Ctitle%253E%253C%2Ftitle%253E%253Cg stroke%3D'none' stroke-width%3D'1' fill%3D'none' fill-rule%3D'evenodd' fill-opacity%3D'0'%253E%253Cg transform%3D'translate(-249.000000%2C -126.000000\&pos_id=img-68yqGk8t-1766808637397)' fill='%23FFFFFF'%3E%3Crect x='249' y='126' width='1' height='1'%3E%3C/rect%3E%3C/g%3E%3C/g%3E%3C/svg%3E)
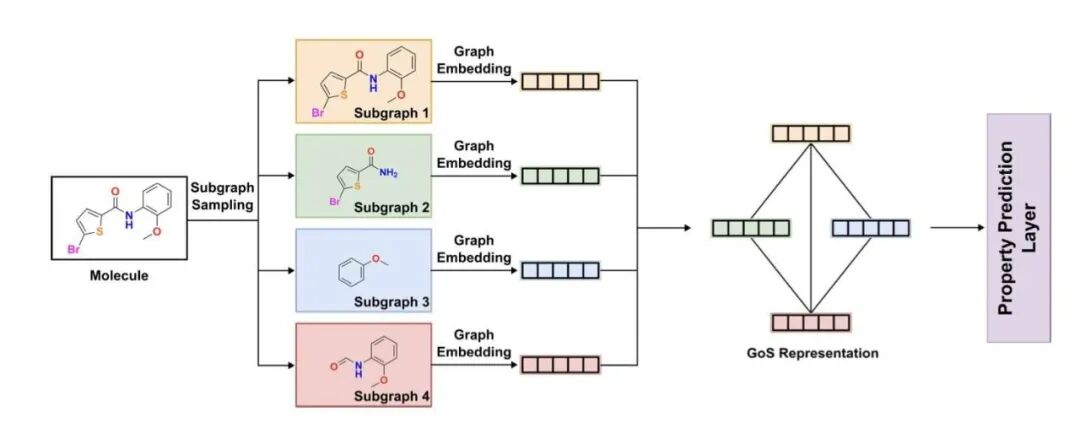
做分子属性预测,我们用了这么多年的图神经网络(Graph Neural Networks, GNNs),但总感觉有个地方不对劲。多数 GNN 模型看分子,就像是把一个精密仪器拆成一堆零件扔进袋子里,只知道里面有几个螺丝、几个齿轮,却完全忽略了这些零件是怎么组装起来的。这种「原子袋」或者「子结构袋」的思路,处理小分子还行,一旦遇到上百个原子的大块头,比如做 OLED 材料或者高分子,就抓瞎了。分子的性质,尤其是电子性质,很大程度上取决于官能团在三维空间里如何「排兵布阵」。
现在,这篇论文提出的 GoMS(Graph of Molecule Substructure)架构,总算有人开始正视这个问题了。
它的思路其实很符合我们化学家的直觉。第一步,先把分子「拆」开。它用的不是暴力的数学切割,而是我们很熟悉的化学工具,比如 BRICS(Breaking of Retrosynthetically Interesting Chemical Substructures)或 RECAP(Retrosynthetic Combinatorial Analysis Procedure)。这些方法本质上是在模拟化学家做逆合成分析时的思考方式,沿着化学上容易断裂的键(比如酰胺键、酯键)切分,得到一个个有化学意义的官能团或结构片段。这样做的好处是双重的:一是得到的子结构是我们能看懂、能解释的;二是可以有效避免把分子切成天文数字般数量的小碎片,控制了计算量。
第二步,也是最关键的一步,是把这些子结构「扶正」,让它们当主角。GoMS 会为每个子结构生成一个嵌入向量(embedding),可以理解为给每个官能团拍了张「身份证照片」,包含了它自身的全部信息。然后,它会构建一个全新的、更高层级的图。在这个新图里,每个节点就是一个子结构,而边则代表了这些子结构之间的拓扑、化学和空间关系。比如,A 官能团和 B 官能团是不是直接相连?它们之间的距离是远是近?这种做法,完美地保留了分子内部的结构层次信息。
这解决了什么核心问题?举个最简单的例子,一个苯环上连着两个取代基,邻位、间位、对位三种异构体的性质天差地别。传统的 GNN 模型可能因为看到的原子和化学键种类数量都一样而犯迷糊,但 GoMS 能清晰地分辨出这三种不同的空间排布,从而给出更准确的预测。
研究者在 OLED 分子和 QM9 这类标准数据集上做了测试。结果不出所料,尤其是在处理那些结构复杂、原子数超过 100 个的工业级大分子时,GoMS 的表现把 ESAN 这类老牌模型甩在了后面。这说明,当分子大到一定程度,官能团之间的长程相互作用和空间构象就成了决定性质的主导因素,而这正是 GoMS 的强项。
他们还做了一个 Graph Transformer 的版本,引入了注意力机制。这就好比给模型配了个广角镜头,让它能同时关注到分子两端距离很远的两个官能团之间的相互影响。这对于理解共轭体系或者有空间位阻效应的分子特别有用。
GoMS 这个工作最大的价值在于,它把化学家的结构思维,也就是这种自下而上、从官能团到整体的层次化视角,成功地融入到了 AI 模型的架构设计里。它不再是让模型去「盲人摸象」,而是先帮它画好了骨架,再让它去填充细节。对于在工业界做材料设计和药物研发的人来说,这种更具化学解释性、又对大分子友好的模型,正是我们需要的。
📜Title: GoMS: Graph of Molecule Substructure Network for Molecule Property Prediction
🌐Paper: https://arxiv.org/abs/2512.12489
--- 完 ---