文章目录
介绍
BayesPrism (Bayesian cell Proportion Reconstruction Inferred using Statistical Marginalization)是一款基于全贝叶斯推断的R包,专为解析**肿瘤微环境(TME)的复杂性而设计。该工具旨在通过统计边缘化方法,利用 单细胞RNA-seq(scRNA-seq)数据作为先验参考,从大量RNA-seq(bulk RNA-seq)**样本中联合估计细胞类型的组成比例以及特定细胞类型的基因表达谱。
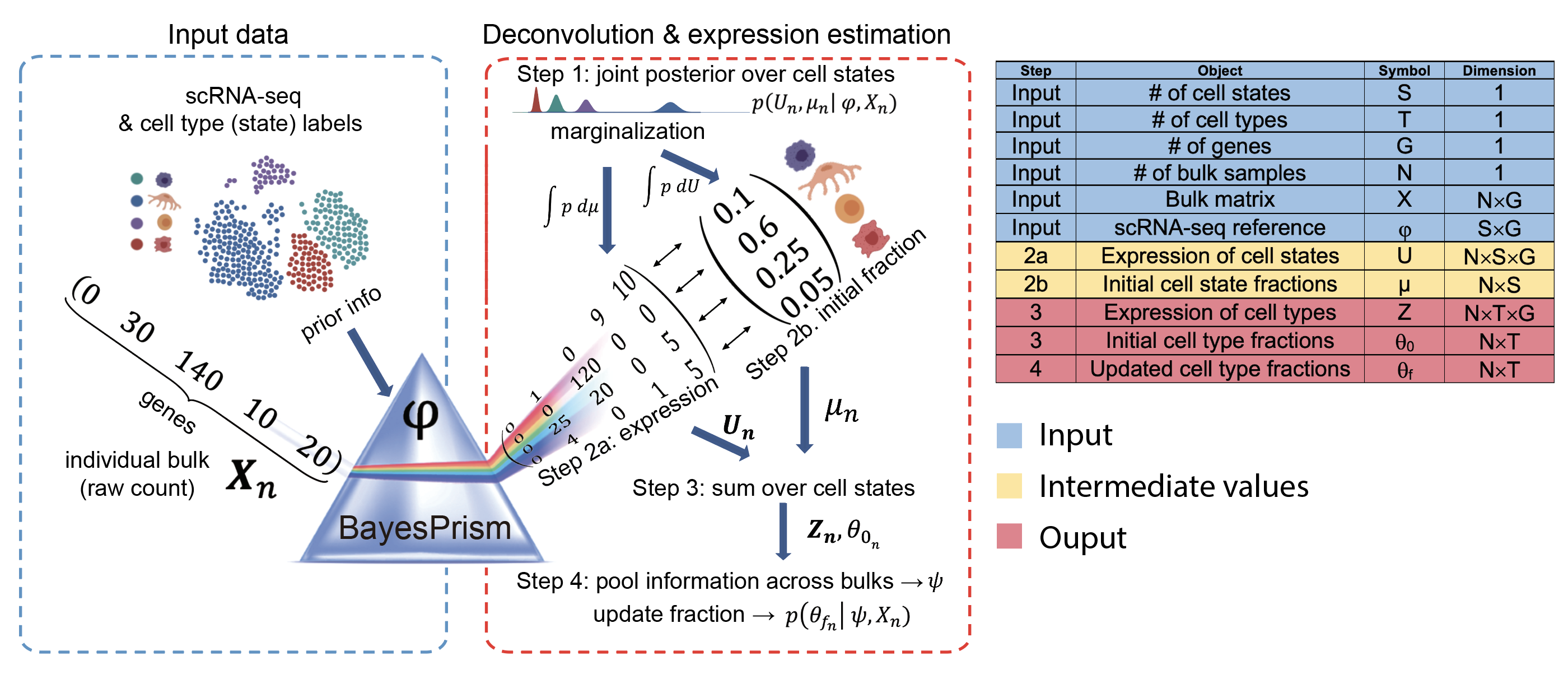
BayesPrism的核心包含两个模块:去卷积模块(Deconvolution module)和嵌入学习模块(Embedding learning module) 。去卷积模块依据scRNA-seq提供的细胞类型特异性表达谱,推断bulk数据中的细胞组分及表达;而嵌入学习模块则使用期望最大化(EM)算法,在非恶性细胞的推断基础上,近似恶性细胞的基因表达程序。最新的V2.2版本支持稀疏矩阵(dgCMatrix)输入,并显著优化了内存效率和运行时间(采用S4对象封装),能够提供细胞类型分数的变异系数(CV)以量化后验分布的不确定性,是研究肿瘤异质性和整合多组学数据的强大工具。
适用条件
- 参考数据集的完备性:BayesPrism假设所有细胞类型的表达谱均已被观测到。因此,建议尽可能使用**完全枚举(complete enumeration)**所有细胞类型的scRNA-seq作为参考。如果遗漏了某种细胞类型,会导致其余细胞类型的比例估计出现膨胀。
- 细胞数量与质量 :建议每个细胞状态(cell state)在参考集中至少包含20个以上的细胞。虽然该工具对测序深度变化具有鲁棒性,但高质量、具有代表性的scRNA-seq参考数据(特别是针对恶性细胞的异质性)能显著提升准确度。
- 避免高相似性干扰 :在构建参考集时,应谨慎包含与肿瘤细胞转录谱高度相似的正常组织细胞(例如在胶质母细胞瘤分析中包含正常星形胶质细胞)。这种相似性可能导致恶性细胞被低估而正常细胞被高估,建议通过留一法(leave-one-out test)测试或利用生物学背景知识进行筛选。
输入数据与功能输出
| 数据/模块类型 | 输入数据描述 | 功能与输出结果 |
|---|---|---|
| 参考数据输入 (Reference) | 单细胞RNA-seq (scRNA-seq) - 原始计数矩阵 (Raw count matrix),V2.2支持稀疏矩阵。 - 细胞类型注释 (cell.type.labels) 及细胞状态注释 (cell.state.labels)。 - 注意:无需预先标准化或对齐基因,包内会自动处理。 | 构建用于去卷积的Prism对象,包含细胞类型特异性的基因表达先验分布。 |
| 混合数据输入 (Mixture) | 大量RNA-seq (Bulk RNA-seq) - 原始计数矩阵 (Raw count matrix)。 - 行名为基因,列名为样本。 - 注意:应排除核糖体、线粒体基因及易受批次效应影响的基因。 | 提供待解构的混合样本表达谱。 |
| 核心功能输出 (Output) | 整合上述两类数据,运行run.prism等函数。 |
1. 细胞类型比例 (Cell Type Fraction):输出初始估计 ( θ 0 \theta_0 θ0) 和更新后的最终估计 ( θ f \theta_f θf)。 2. 特定细胞类型基因表达 (Cell Type-Specific Expression):从Bulk数据中推断出的各类细胞的基因表达谱。 3. 不确定性量化 :输出细胞类型分数的变异系数 (CV)。 4. 肿瘤基因程序嵌入:恶性细胞的线性组合表达特征。 |
2个Demo
使用 BayesPrism 进行恶性细胞表达的嵌入学习
BayesPrism 包含一个可选的嵌入学习模块,用于识别去除肿瘤浸润的非恶性细胞后,在批量 RNA-seq 样本中常见的基因表达模式。
r
# 1. 加载包与数据
library(BayesPrism)
library(NMF) # 需要 NMF 包来进行 rank 扫描
load("bp.res.rdata") # 加载之前的 BayesPrism 去卷积结果 (bp.res)
# 2. 选择恶性程序的数量 K
# 提取更新后的恶性细胞表达谱 (归一化)
Z.tum.norm <- t(bp.res@reference.update@psi_mal)
# 使用 NMF 扫描 K 值 (此处示例为 2 到 12)
# 注意:这步计算量较大,耗时较长
estim.Z.tum.norm <- nmf(Z.tum.norm, rank=2:12, seed=123456)
# 可视化各项指标与共识图以辅助选择 K 值
plot(estim.Z.tum.norm)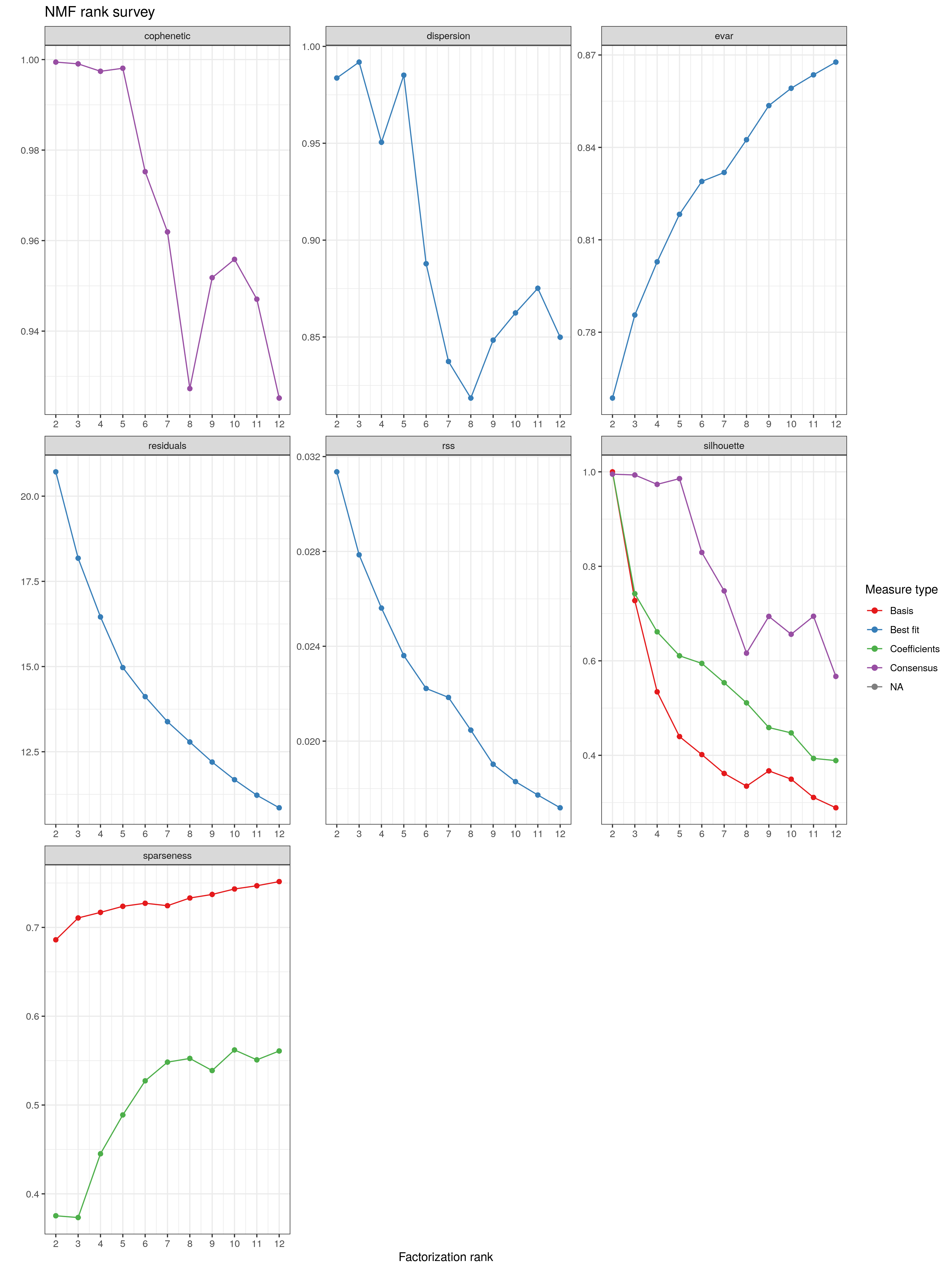
r
consensusmap(estim.Z.tum.norm, labCol=NA, labRow=NA)
r
# 3. 运行嵌入式学习 (Embedding Learning)
# 假设根据上一步选择了 K = 5
ebd.res <- learn.embedding.nmf(
bp = bp.res, # 输入必须是在 tumor mode 下运行的 BayesPrism 结果
K = 5, # 选定的 K 值
cycle = 20, # EM 迭代次数 (通常约 50 次收敛,演示设为 20)
compute.elbo = T # 计算 ELBO 以监控收敛情况
)
# 查看结果结构
str(ebd.res)
# 绘制 ELBO 曲线检查收敛性
plot(ebd.res$elbo, xlab="EM cycle", ylab="-ELBO", type="l")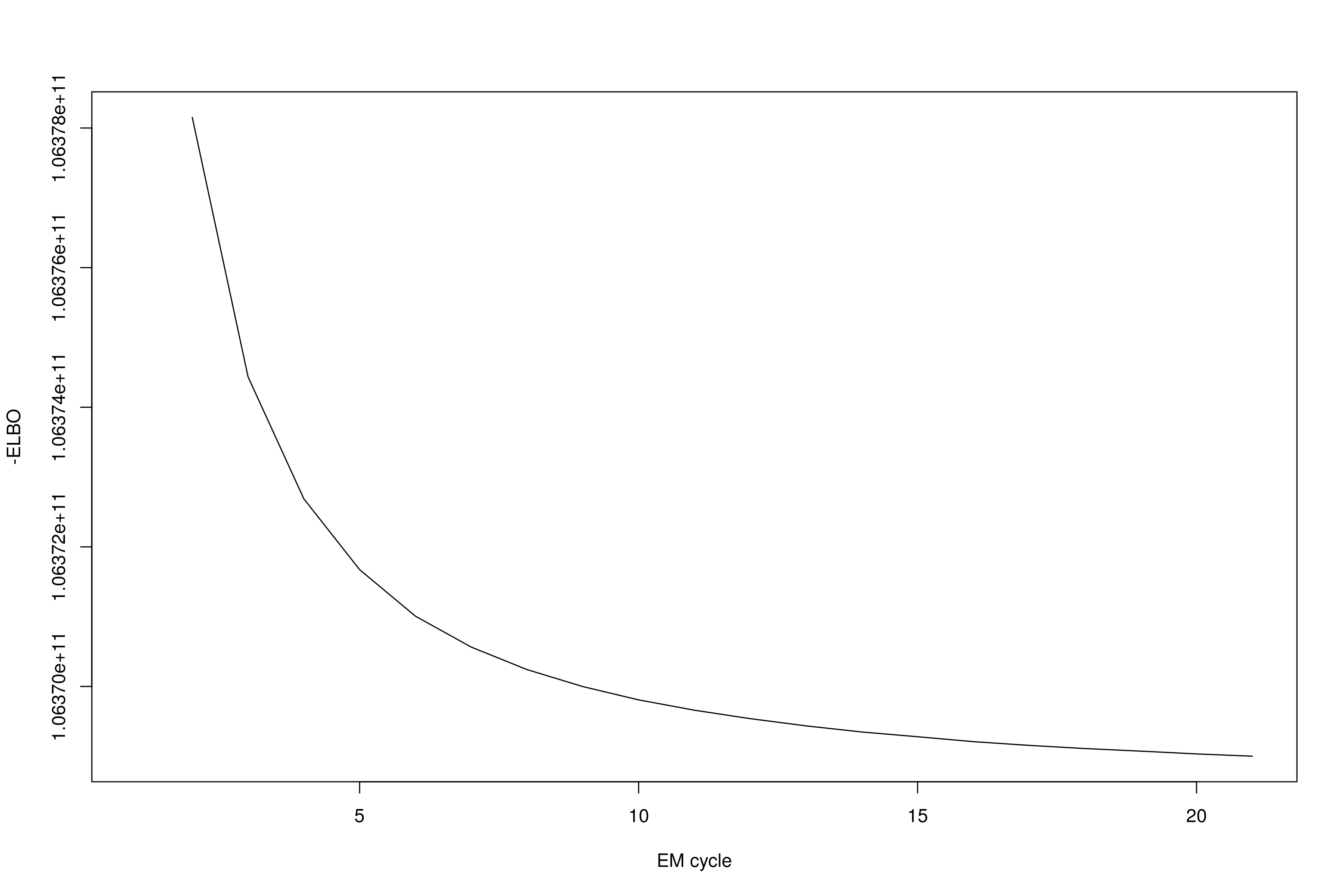
r
# 4. (可选) 继续增加迭代次数
# 如果 ELBO 未收敛,可在上一步结果基础上继续运行
ebd.res.2 <- learn.embedding(
bp = bp.res,
cycle = 10, # 额外增加 10 次循环
EM.res = ebd.res, # 传入上一步的运行结果
compute.elbo = T
)
# 绘制合并后的 ELBO 曲线
plot(c(ebd.res$elbo, ebd.res.2$elbo[-1]), xlab="EM cycle", ylab="-ELBO", type="l")
# 5. (可选) 使用自定义先验运行
# 如果有特定的生物学先验知识 (eta_prior)
# ebd.res.myEta <- learn.embedding(
# bp = bp.res,
# eta_prior = my.eta, # 用户提供的 K-by-G 矩阵 (raw count scale)
# cycle = 50,
# compute.elbo = T
# )
使用 BayesPrism 进行Bulk RNA-seq 反卷积
BayesPrism 利用从匹配或相似组织类型中采集的样本的单细胞 RNA 测序 (scRNA-seq) 数据,对批量 RNA 测序(以及空间转录组学)进行细胞类型和基因表达反卷积。它将 scRNA-seq 数据视为先验信息,并进行估计 P ( θ , Z ∣ X , ϕ ) P(\theta, Z | X, \phi) P(θ,Z∣X,ϕ),即在参考 ϕ \phi ϕ 和每个观测到的批量样本 X X X 的条件下,联合估计细胞类型比例的后验分布 θ \theta θ 以及细胞类型特异性基因表达 Z Z Z。
- 示例数据:
- Github:https://dreg.dnasequence.org/
- 登不了Github的小伙伴后台回复【20251230bayesprism 】下载包
r
suppressWarnings(library(BayesPrism))
load("../tutorial.dat/tutorial.gbm.rdata")
ls()
# [1] "bk.dat" "cell.state.labels" "cell.type.labels" "sc.dat"
# 细胞状态相关性热图
plot.cor.phi(input=sc.dat,
input.labels=cell.state.labels,
title="cell state correlation",
cexRow=0.2, cexCol=0.2,
margins=c(2,2))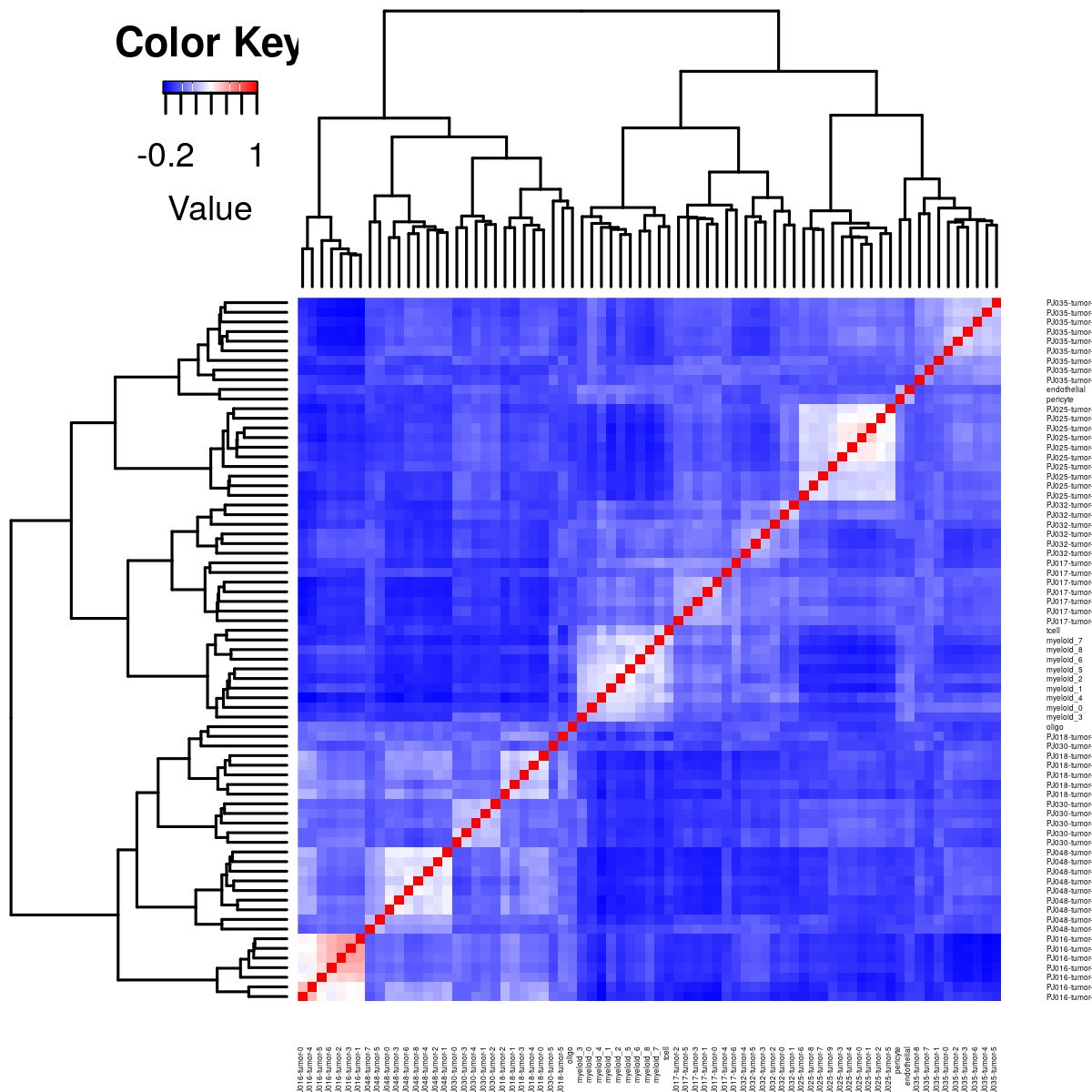
r
# 细胞类型相关性热图
plot.cor.phi(input=sc.dat,
input.labels=cell.type.labels,
title="cell type correlation",
cexRow=0.5, cexCol=0.5)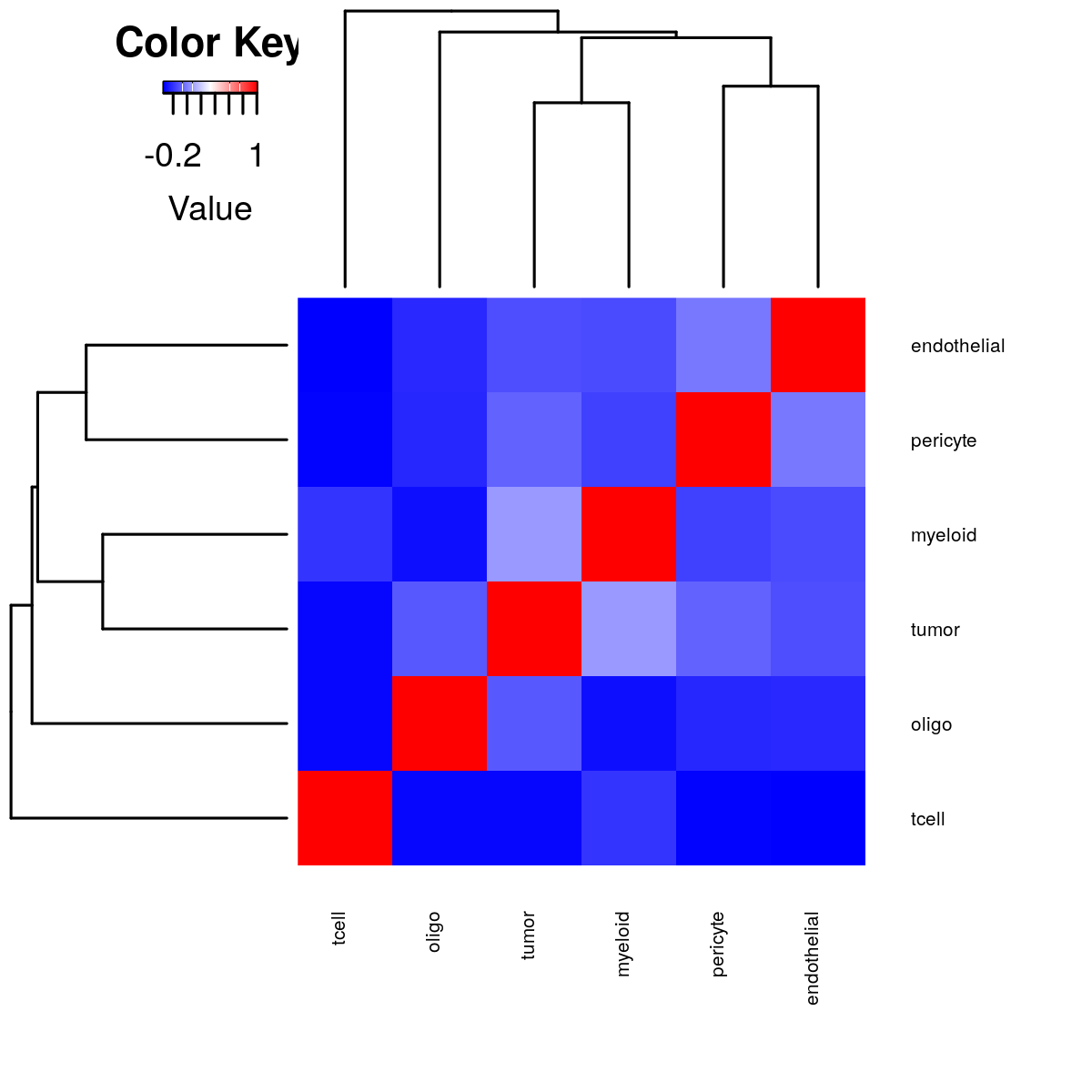
r
# scRNA异常基因分布图
sc.stat <- plot.scRNA.outlier(input=sc.dat,
cell.type.labels=cell.type.labels,
species="hs",
return.raw=TRUE)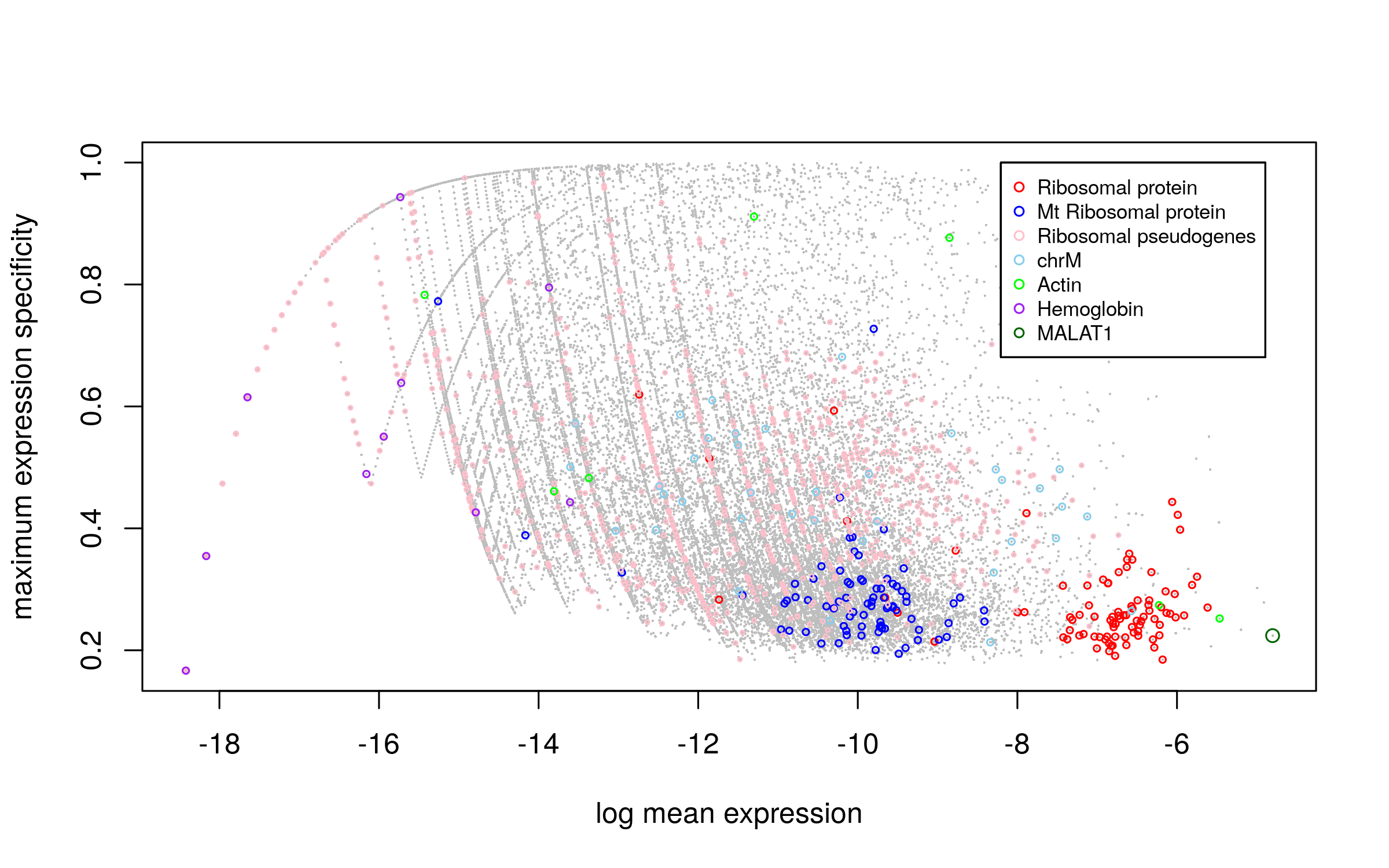
r
# Bulk RNA异常基因分布图
bk.stat <- plot.bulk.outlier(bulk.input=bk.dat,
sc.input=sc.dat,
cell.type.labels=cell.type.labels,
species="hs",
return.raw=TRUE)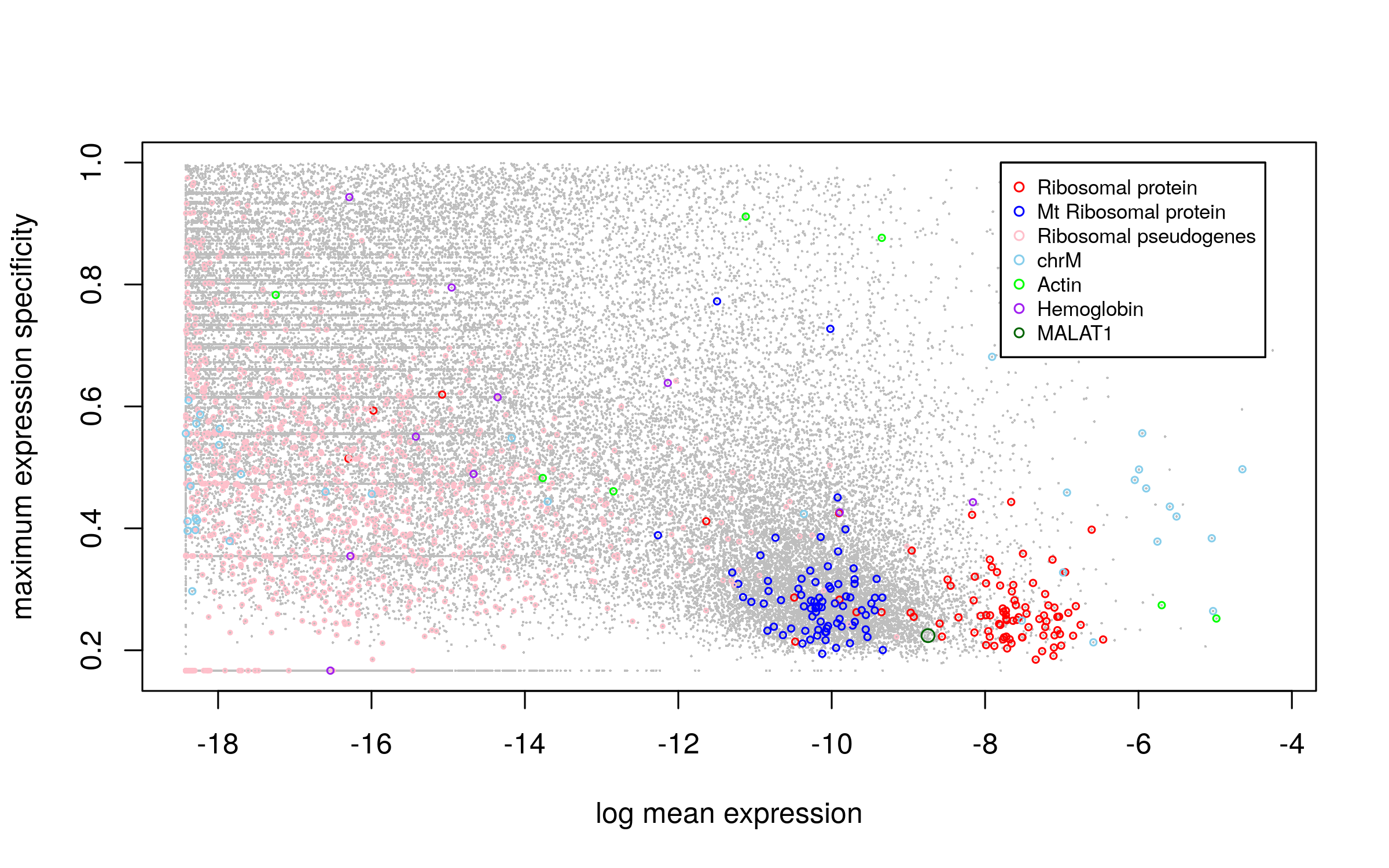
r
sc.dat.filtered <- cleanup.genes(input=sc.dat,
input.type="count.matrix",
species="hs",
gene.group=c("Rb","Mrp","other_Rb","chrM","MALAT1","chrX","chrY"),
exp.cells=5)
# Bulk与SC表达一致性散点图
plot.bulk.vs.sc(sc.input = sc.dat.filtered,
bulk.input = bk.dat)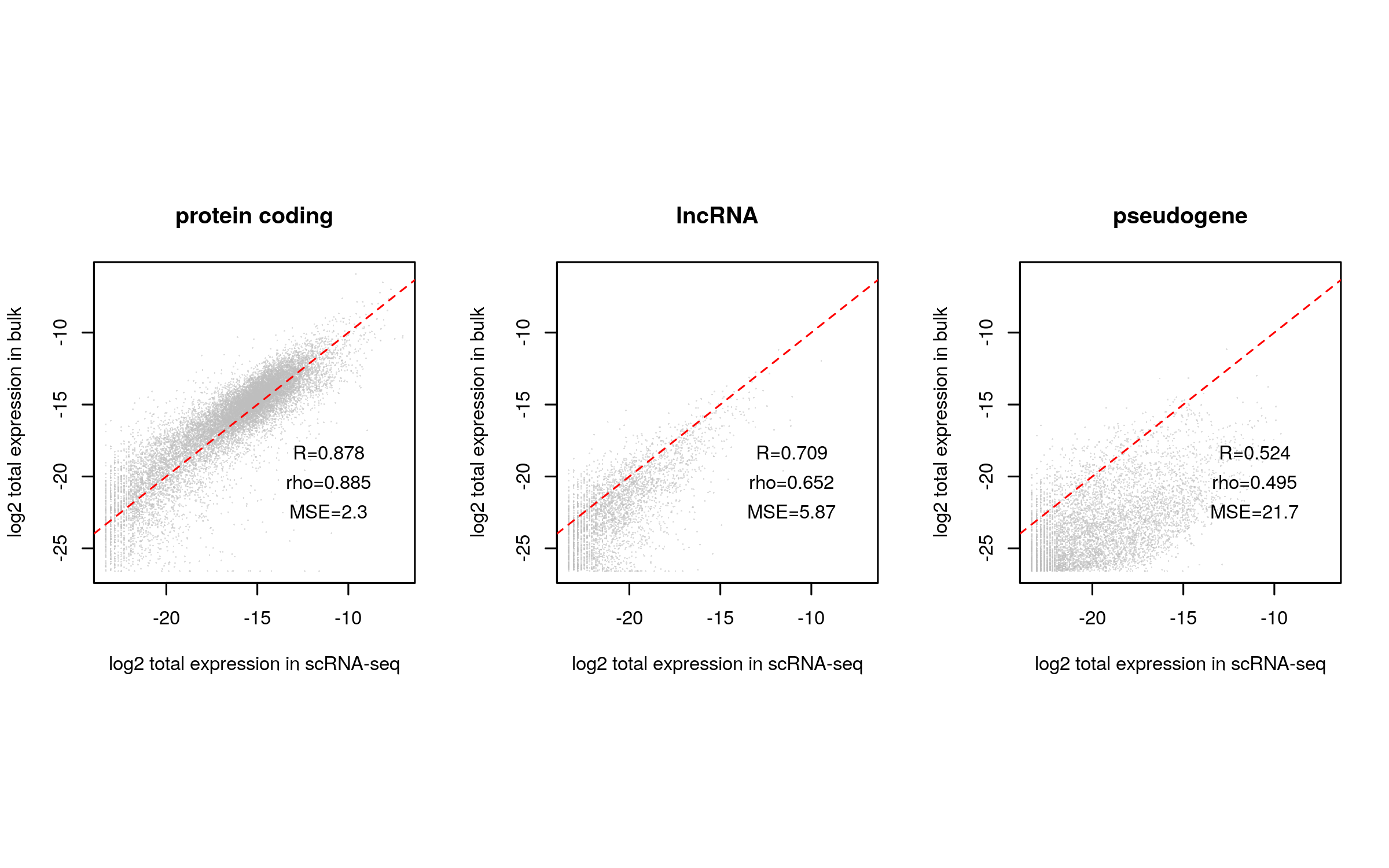
r
# 只保留蛋白质编码基因(可选)
sc.dat.filtered.pc <- select.gene.type(sc.dat.filtered,
gene.type = "protein_coding")
# 对不同细胞类型中的细胞状态进行成对 t 检验
diff.exp.stat <- get.exp.stat(sc.dat=sc.dat[,colSums(sc.dat>0)>3],
cell.type.labels=cell.type.labels,
cell.state.labels=cell.state.labels,
pseudo.count=0.1,
cell.count.cutoff=50,
n.cores=1)
# 筛选特征基因
sc.dat.filtered.pc.sig <- select.marker(sc.dat=sc.dat.filtered.pc,
stat=diff.exp.stat,
pval.max=0.01,
lfc.min=0.1)
myPrism <- new.prism(reference=sc.dat.filtered.pc, # 或使用筛选过特征基因的 sc.dat.filtered.pc.sig
mixture=bk.dat,
input.type="count.matrix",
cell.type.labels = cell.type.labels,
cell.state.labels = cell.state.labels,
key="tumor",
outlier.cut=0.01,
outlier.fraction=0.1)
# 运行 BayesPrism
bp.res <- run.prism(prism = myPrism, n.cores=50)
# 获取最终更新后的后验均值
theta <- get.fraction(bp=bp.res,
which.theta="final",
state.or.type="type")
head(theta)
# 提取细胞类型分数的变异系数 (CV) 以评估不确定性
theta.cv <- bp.res@posterior.theta_f@theta.cv
head(theta.cv)
# 提取特定细胞类型的基因表达矩阵 Z(例如肿瘤细胞)
Z.tumor <- get.exp(bp=bp.res,
state.or.type="type",
cell.name="tumor")
head(t(Z.tumor[1:5,]))
save(bp.res, file="bp.res.rdata")- 简易生信分析问题可免费解答,复杂问题付费咨询;欢迎投稿需要复现的文献图表,团队可整理和分享一份案例交流学习。
- 期待学术合作者 加入团队,磨合后有数据有课题有契机的可合作冲子刊和正刊,另外筹备SCI期刊编委会。
- 承接单细胞空转真核转录组等多组学测序服务(寻因平台 ) ,欢迎各大医院或课题组咨询,关注6个月以上给予绝对低于市场价的粉丝价。
- 欢迎扩列交流学习聊职业规划等 ,健谈和爱交朋友,同时非常欢迎生信、AI多模态领域高手加入团队合作储备人才。