KRAS作为原癌基因家族的核心成员之一,编码小GTP酶。正常情况下,KRAS蛋白通过结合GTP(激活态)或 GDP(失活态)进行状态的转换,调控细胞增殖、分化、凋亡及迁移等关键生理过程。当KRAS发生突变时,其GTP水解活性受阻,蛋白持续处于GTP结合的激活状态,导致下游信号通路过度激活,驱动细胞恶性转化,最终引发细胞异常增殖,并与细胞的侵袭性、转移性及不良表型密切相关。由于KRAS蛋白表面光滑,缺少明确的结合口袋,且KRAS与底物GTP的亲和力极高,难以竞争,长期以来缺少对应的抑制剂,直至2013年针对KRAS突变的共价抑制剂被发现,才打破这一困境。
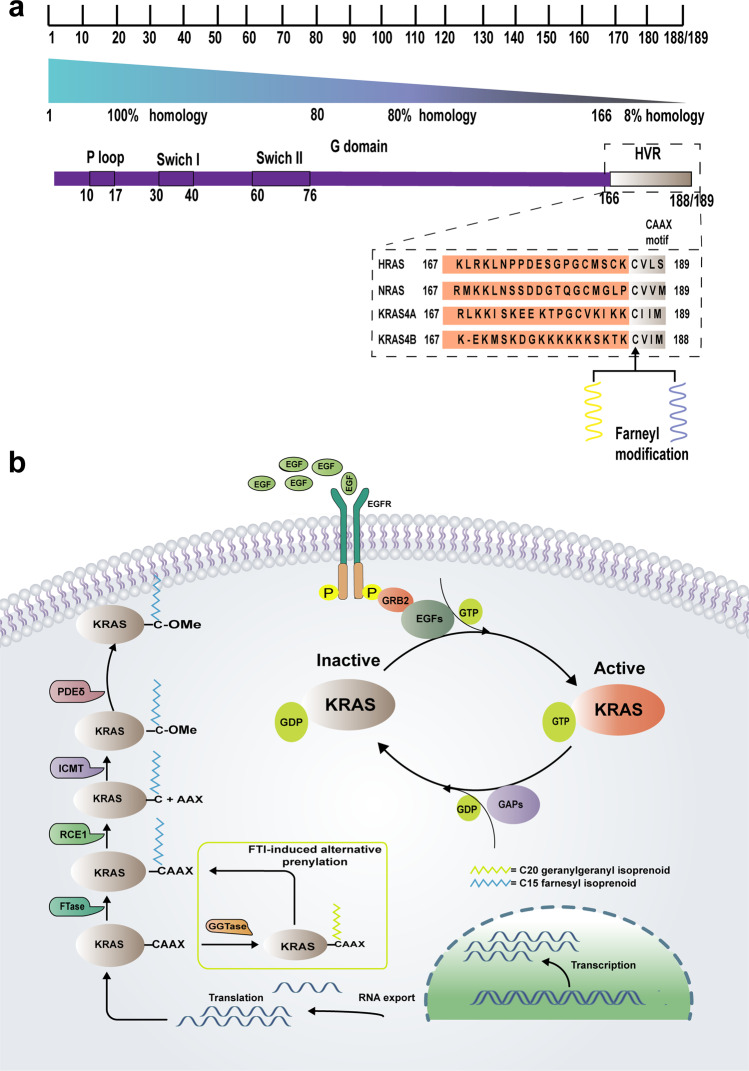
图 1.
一、KRAS G12C 抑制剂
Sotorasib(AbMole,AMG 510,M9356)作为首个进入系统性研究阶段的KRAS共价抑制剂,能通过丙烯酰胺基团与KRASG12C突变体的12位Cys残基形成共价键,特异性结合KRAS突变体的Switch II口袋,把KRAS蛋白锁定在失活构象。体外实验表明,Sotorasib(AbMole,AMG 510,M9356)能显著抑制KRAS突变细胞的增殖并诱导凋亡,对野生型KRAS无明显影响,在非小细胞肺癌(NSCLC)的研究中有着重要应用。Sotorasib(CAS No.:2296729-00-3)对A549细胞的IC50为36.5 μM1
Adagrasib(MRTX849,AbMole,M9428)作为高效KRASG12C共价抑制剂,分子结构与Sotorasib(AbMole,AMG 510)相近,对KRAS突变体结合亲和力较高。体外实验证实,其在纳摩尔浓度下即可抑制KRAS突变细胞的增殖,对多种来源的KRASG12C突变细胞均表现出显著的抑制活性。Adagrasib(CAS No.:2326521-71-3)对AGS细胞的IC50为1.4 μM,对A549细胞的IC50为12.9 μM,对MIA PaCa-2的IC50则低至4 nM。
RMC-7977(AbMole,M54858)是一种广谱、高选择性的RAS (ON)多选择性抑制剂,RMC-7977的核心作用机制是靶向RAS蛋白的GTP结合活性状态(即RASON构象),能同时抑制突变型和野生型KRAS、NRAS及HRAS的活性,从而阻断下游MAPK信号通路(RAS-RAF-MEK-ERK)的持续激活2。在多种肿瘤模型中,RMC-7977通过抑制RAS信号传导,诱导细胞周期阻滞和凋亡。例如,在KRAS突变型结直肠癌细胞中,RMC-7977能有效阻断ERK磷酸化,抑制增殖并诱导部分细胞的凋亡3。值得注意的是,RMC-7977还能重塑肿瘤免疫微环境。在PDAC和黑色素瘤模型中,RMC-7977(CAS No.:2765082-12-8)能减少免疫抑制性髓系细胞的浸润,同时增加CD4+和CD8+ T细胞以及常规树突状细胞的浸润,从而触发免疫系统介导的肿瘤消退4。
Divarasib(GDC-6036,AbMole,M11442)是新一代的KRAS共价抑制剂,Divarasib(CAS No.:2417987-45-0)依赖其亲电性基团(如丙烯酰胺)与KRASG12C蛋白中突变的Cys12形成不可逆的共价加合物,实现对KRASG12C的持久抑制。Divarasib不仅具有持续且显著的共价结合能力,还包含较强的非共价结合成分。Divarasib在非小细胞肺癌小鼠抑制模型中,10-100 mg/kg的剂量有效抑制了肿瘤的生长5。
Daraxonrasib(RMC-6236,AbMole,M40832)是一种首创的三重复合泛RAS抑制剂。Daraxonrasib(CAS No.:2765081-21-6)通过与细胞内丰富的免疫亲和蛋白亲环素A(Cyclophilin A, CypA)高亲和力结合(Kd=55.3 nmol/L),形成二元复合物;该复合物随后与处于活性GTP结合状态的KRAS蛋白(包括KRASG12D、KRASG12V和野生型KRAS,Kd分别为131 nmol/L、364 nmol/L和154 nmol/L)结合,形成稳定的三元复合物(CypA:Daraxonrasib:KRAS-GTP),通过空间位阻阻断RAS与下游效应蛋白(如RAF)的相互作用,从而抑制MAPK信号通路,最终抑制肿瘤细胞增殖。值得注意的是,Daraxonrasib(RMC-6236,AbMole,M40832)属于RAS(ON)多选择性抑制剂,不仅对KRAS G12C突变有效,还对多种其他KRAS突变(如G12D、G12V等)甚至野生型RAS具有广谱抑制活性6。
Opnurasib(JDQ-443,AbMole,M11016)是一种不可逆的共价KRASG12C抑制剂,与Sotorasib 和Adagrasib 不同,Opnurasib(CAS No.:2653994-08-0)同时具有靶向GDP和GTP结合态的KRASG12C的能力,因此Opnurasib能抑制处于激活状态下的KRASG12C。实验结果表明,Opnurasib(JDQ-443,AbMole,M11016)能有效抑制RAS通路下游蛋白ERK的激活,并在非小细胞肺癌和结肠癌的细胞与动物模型中表现出抗肿瘤潜力。
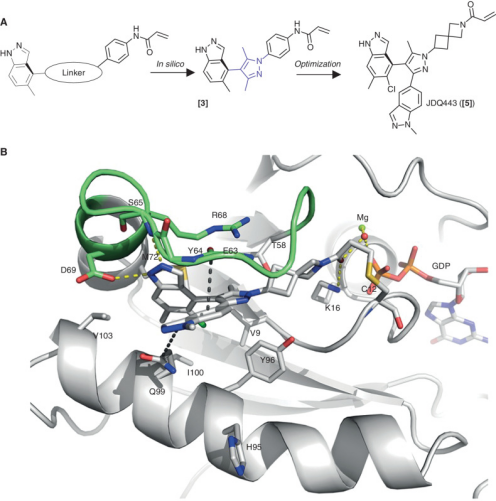
图 2. Design of JDQ443 and its binding mode to KRASG12C6
ARS-1620(AbMole,M9045)是一种早期开发的KRAS共价抑制剂,通过丙烯酰胺基团与KRAS的Cys12共价结合,抑制KRAS突变体活性。另外,ARS-853(AbMole,M9235)也是一种靶向KRASG12C突变体的共价抑制剂。高分辨率晶体结构分析证实,ARS-853(CAS No.:1629268-00-3)能共价标记KRAS的Cys12并占据S-IIP,导致α2螺旋显著旋转,同时与α3螺旋相互作用,从而稳定KRASG12C的无活性GDP结合状态7。在细胞实验中,ARS-853在H358肺癌细胞中的IC₅₀为1.6 μM(6小时处理),能剂量依赖性地减少GTP结合型KRAS的水平,抑制下游效应子CRAF、ERK和AKT的磷酸化及相互作用,并诱导细胞凋亡8。
MRTX-1257(AbMole,M14054)是一款选择性共价KRASG12C抑制剂,能靶向GDP结合态KRASG12C的Switch II口袋,在蛋白试验中,3 μM的MRTX-1257(CAS No.:2206736-04-9)在5分钟内对KRASG12C的修饰率高达59%;MRTX-1257在H358细胞中IC₅₀为0.9 nM;MRTX-1257在小鼠MIA PaCa-2 G12C移植模型中,小鼠口服1、3、10、30、100 mg/kg的MRTX-1257(每日一次连续30天),所有剂量组均不同程度地抑制了肿瘤生长。
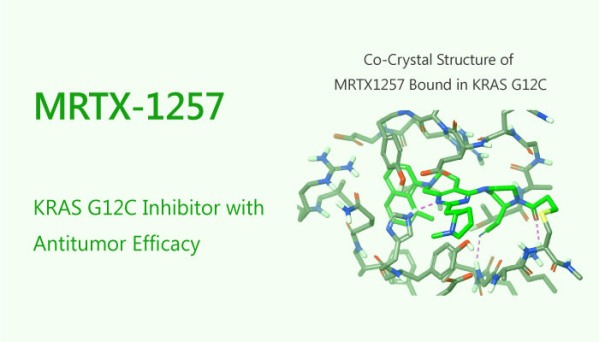
图 3. MRTX-1257 is a KRAS G12C Inhibitor with Antitumor Efficacy
BI-2493(AbMole,M25594)是一种非共价泛KRAS抑制剂,能优先结合KRAS失活态(GDP结合态),通过稳定KRAS在失活态阻止效应蛋白募集,BI-2493(CAS No. : 2937344-16-4)对KRAS的选择性源于BI-2493自身结构与G12、G13和Q61三个关键残基形成的独特氢键9。
LC-2(AbMole,M10385)是首个可降解内源性KRASG12C的PROTAC,由KRAS抑制剂MRTX849 与VHL E3泛素连接酶配体偶联而成;LC-2(CAS No.:2502156-03-6)在NCI-H2030细胞中DC₅₀为0.59±0.20 μM,最大降解率约80%;在MIA PaCa-2、SW1573、NCI-H23和NCI-H358细胞中DC₅₀分别为0.32±0.08、0.76±0.30、0.25±0.08和0.52±0.30 μM;LC-2通过形成三元复合物(KRAS-PROTAC-VHL)诱导KRASG12C泛素化和蛋白酶体依赖性降解,抑制下游ERK信号传导10。
Elironrasib(RMC-6291,AbMole,M25612)是一种RAS(ON) G12C选择性抑制剂,靶向结合GTP的KRASG12C,形成共价三元复合物(KRAS-抑制剂-亲环蛋白A),通过空间位阻阻断RAS效应子结合;在KRAS G12C突变H358细胞中抑制ERK信号并诱导凋亡,IC₅₀低至0.11 nM;Elironrasib在小鼠肿瘤模型中200 mg/kg的剂量口服每日一次,连续60天后显著抑制肿瘤生长11。
ASP2453(AbMole,M21220)是一种新型的KRASG12C共价抑制剂,能与KRASG12C蛋白共价结合;ASP2453(CAS No.:2241719-73-1)在NCI-H1373细胞中以1 nM作为起始浓度,结果显示其剂量依赖性抑制了KRAS-GTP、p-Akt和p-ERK的水平;ASP2453在MIA PaCa-2细胞中3 nM的浓度抑制KRAS-GTP、p-ERK和p-S6的水平;ASP2453还在NCI-H1373异种移植模型(小鼠)中以5 mg/kg(口服给药)抑制肿瘤生长,其中10 mg/kg和30 mg/kg分别诱导47%和86%肿瘤退缩12。
BBO-8520(AbMole,M55031)是一种首创的共价双抑制剂,同时靶向GTP结合和GDP结合状态的KRASG12C;结合于Switch-II/Helix3口袋,共价修饰KRAS的半胱氨酸,并阻断KRASG12C(ON)与下游效应蛋白的结合;BBO-8520(CAS No.:2893809-51-1)在MIA PaCa-2和SW1473细胞中20 nM处理30分钟即能显示出快速的pERK抑制并持续至24小时;BBO-8520在MIA PaCa-2异种移植(小鼠)模型中30 mg/kg的剂量诱导明显的肿瘤退缩13。
二、 KRAS G12D 抑制剂
KRAS G12D占KRAS突变肿瘤中的30%左右,在胰腺导管腺癌(PDAC)的KRAS突变中占比则高达40%。然而,与KRAS G12C不同,KRAS G12D突变缺乏易于靶向的半胱氨酸残基,取而代之的是天冬氨酸残基(Asp12)。MRTX1133(AbMole,M10593)是第一款非共价、强效和选择性KRASG12D突变体抑制剂。MRTX1133(CAS No.:2621928-55-8)与KRASG12D具有较高的亲和力,可同时抑制激活和失活状态下的KRASG12D。MRTX1133分子中的哌嗪基取代基可与Asp12的羧基形成强静电相互作用;中心的吡啶-嘧啶支架与Tyr96产生π-π堆叠;而萘环部分则与疏水口袋结合,提供额外的结合力。这些相互作用共同稳定了MRTX1133 与KRASG12D的结合,从而有效阻断KRAS的激活。研究数据显示,MRTX1133对表达KRASG12D的肿瘤细胞的IC50范围多为1.5-50 nM14,对胰腺癌细胞系(AsPc-1)和胃癌细胞系(AGS)的半数抑制浓度(IC50)分别为1.4 nM和7.9 nM14。
Zoldonrasib(RMC-9805,AbMole,M55018)是一种KRAS(ON)G12D选择性抑制剂,可特异性结合处于激活态的KRASG12D突变蛋白,Zoldonrasib(CAS No.:2922732-54-3)通过氮丙啶弹头共价结合天冬氨酸,阻断其与下游效应蛋白结合。Zoldonrasib在多种KRAS突变细胞模型中抑制效果显著,可有效抑制RAS/MAPK信号通路。Zoldonrasib对乳腺癌细胞(KRASG12D突变)的半数抑制浓度(IC50)为43.51 µM,对肺癌细胞的IC50为18.78 µM15。Zoldonrasib(RMC-9805)在三阴性乳腺癌异种移植小鼠模型中,经口服给药(剂量为5 mg/kg),每日一次,显示出显著的抗肿瘤效果16。
三、 KRAS互作抑制剂
BI-3406 (AbMole,M9801)是一种高效的 KRAS-SOS1 抑制剂,能抑制 KRAS与SOS1之间相互作用。SOS1(Son of Sevenless 1)是一种鸟苷酸交换因子(GEF),其核心功能是催化KRAS蛋白上的GDP向GTP的交换,将KRAS从失活状态(KRAS-GDP)转化为激活状态(KRAS-GTP),从而启动下游信号传导。这一过程对细胞增殖、分化及存活至关重要,已经在多种肿瘤中发现了KRAS-SOS1的异常变化。BI-3406(CAS No.:2230836-55-0)通过结合SOS1的催化结构域,有效阻断SOS1与KRAS之间的蛋白-蛋白相互作用,从而抑制RAS从GDP向GTP的交换,减少活化的GTP-RAS水平。BI-3406对SOS1-KRAS相互作用的IC50为3.9 nM(体外无细胞系统)。BI-3406对A549、ASPC1、K562等细胞的IC50分别为106 nM、64.4 nM、1.422 μM17。
BAY-293(AbMole,M9359)也是一种高效的SOS1抑制剂,破坏KRAS-SOS1相互作用,细胞游离IC₅₀为21 nM;在HeLa细胞中阻断RAS活化的IC₅₀为410 nM,在K562细胞中60分钟孵育后有效降低pERK(KRAS下游效应蛋白)水平(IC₅₀为180 nM);BAY-293(CAS No.:2244904-70-7)在野生型KRAS细胞系(K562、MOLM-13)中IC₅₀约1 μM,在KRAS G12C突变细胞系(NCI-H358、Calu-1)中IC₅₀约3 μM;有文献表明:BAY-293与ARS-853(KRAS G12C共价抑制剂)联合显示协同效应18。
Deltarasin(AbMole,M3394)是首个小分子KRAS-PDEδ相互作用抑制剂,能结合PDEδ的法尼基结合腔,Kd为41 nM;抑制PDEδ-KRAS相互作用,干扰KRAS在质膜上的定位,进而抑制下游通路信号和癌细胞增殖;在Panc-Tu-I异种移植裸鼠模型中,10和15 mg/kg的Deltarasin处理9天,结果显示Deltarasin(CAS No.:1440898-61-2)能以剂量依赖的方式抑制细胞生长、减少肿瘤体积19。
四、 KRAS膜定位抑制剂
Fendiline(AbMole,M5640)是一种L型钙通道阻滞剂,能特异性抑制K-Ras质膜定位而不影响H-Ras和N-Ras,Fendiline(CAS No.:13636-18-5)通过显著减少K-Ras簇的形成并将其从质膜重新分布至内质网、高尔基体、内吞体和细胞质,通过这种方式来间接地阻断K-Ras信号通路对外界信号的接收或非特异性激活20。
FGTI-2734(AbMole,M28036)是RAS C末端法尼基转移酶 (FT) 和香叶烯基转移酶-1 (GGT-1) 的抑制剂,对FT和GGT-1的IC₅₀分别为250 nM和520 nM;FGTI-2734(CAS No.:1247018-19-4)能阻断KRAS膜定位以解决肿瘤细胞对KRAS抑制剂的耐受性问题;FGTI-2734(30 μM)在MiaPaCa2、L3.6pl和Calu6细胞中处理72小时后诱导Caspase-3和PARP裂解,并抑制HDJ2、RAP1A、KRAS和NRAS的蛋白质异戊烯化;FGTI-2734在小鼠中腹腔注射100 mg/kg/天,连续处理25天后显著抑制KRAS突变型肿瘤的生长21。
范例详解
iScience. 2023 Jan 28;26(2):106080.
蛋白质组学国家重点实验室、中科院植物研究所的实验人员在上述文章中探究了KRAS G12C 共价抑制剂AMG510的蛋白修饰谱,并通过开发两种分析方法(直接Profiling和基于泛 AMG510抗体的肽段免疫沉淀),系统性鉴定了AMG510的靶向及脱靶修饰位点,揭示了其潜在的生物学效应和毒理学意义。上述论文的研究核心---KRAS G12C 共价抑制剂AMG510(Sotorasib,AbMole,M9356)由AbMole提供。此外,实验人员还使用了AbMole的MRTX849(Adagrasib,AbMole,M9428),另一种与AMG510结构相似的 KRAS G12C共价抑制剂,以验证泛 AMG510 抗体的靶向性,即该抗体仅识别 AMG510 修饰的肽段,不识别 MRTX849 修饰或未修饰的肽段22。
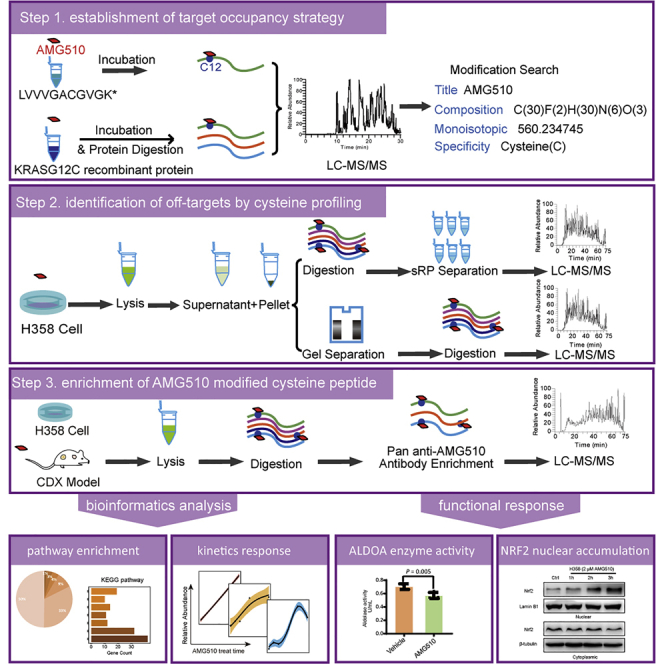
图 4. Global profiling of AMG510 modified proteins identified tumor suppressor KEAP1 as an off-target22
参考文献及鸣谢
1 Yang, N.; Fan, Z.; Sun, S.; et al. Discovery of highly potent and selective KRAS(G12C) degraders by VHL-recruiting PROTACs for the treatment of tumors with KRAS(G12C)-Mutation. European journal of medicinal chemistry 2023 , 261, 115857.
2 Popescu, B.; Jones, M. F.; Piao, M.; et al. Multiselective RAS(ON) inhibition targets oncogenic RAS and overcomes RAS-mediated resistance to FLT3i and BCL2i in AML. Blood 2026 , 147 (3), 276-289.
3 Aust, O.; Thiel, M. R. T.; Blanc, E.; et al. Reporter-based screening identifies RAS-RAF mutations as drivers of resistance to active-state RAS inhibitors in colorectal cancer. Science signaling 2025 , 18 (896), eadr3738.
4 Anastacio Da Costa Carvalho, L.; Tovbis Shifrin, N.; Phadke, M. S.; et al. RAS(ON) Multiselective Inhibition Drives Antitumor Immunity in Preclinical Models of NRAS-Mutant Melanoma. Cancer immunology research 2026 , 14 (1), 90-106.
5 Meng, L.; Chan, E. W.; Ng, C.; et al. Assessment of KRAS G12C Target Engagement by a Covalent Inhibitor in Tumor Biopsies Using an Ultra-Sensitive Immunoaffinity 2D-LC-MS/MS Approach. Analytical chemistry 2022 , 94 (37), 12927-12933.
6 Cregg, J.; Edwards, A. V.; Chang, S.; et al. Discovery of Daraxonrasib (RMC-6236), a Potent and Orally Bioavailable RAS(ON) Multi-selective, Noncovalent Tri-complex Inhibitor for the Treatment of Patients with Multiple RAS-Addicted Cancers. Journal of medicinal chemistry 2025 , 68 (6), 6064-6083.
7 Lu, J.; Harrison, R. A.; Li, L.; et al. KRAS G12C Drug Development: Discrimination between Switch II Pocket Configurations Using Hydrogen/Deuterium-Exchange Mass Spectrometry. Structure (London, England : 1993) 2017 , 25 (9), 1442-1448.e1443.
8 Shipman, L. Signalling: Putting the brakes on KRAS-G12C nucleotide cycling. Nat Rev Cancer 2016 , 16 (3), 127.
9 Tedeschi, A.; Schischlik, F.; Rocchetti, F.; et al. Pan-KRAS Inhibitors BI-2493 and BI-2865 Display Potent Antitumor Activity in Tumors with KRAS Wild-type Allele Amplification. Molecular cancer therapeutics 2025 , 24 (4), 550-562.
10 Bond, M. J.; Chu, L.; Nalawansha, D. A.; et al. Targeted Degradation of Oncogenic KRAS(G12C) by VHL-Recruiting PROTACs. ACS central science 2020 , 6 (8), 1367-1375.
11 Drizyte-Miller, K.; Talabi, T.; Somasundaram, A.; et al. KRAS: the Achilles' heel of pancreas cancer biology. The Journal of clinical investigation 2025 , 135 (16).
12 Nakayama, A.; Nagashima, T.; Nishizono, Y.; et al. Characterisation of a novel KRAS G12C inhibitor ASP2453 that shows potent anti-tumour activity in KRAS G12C-mutated preclinical models. Br J Cancer 2022 , 126 (5), 744-753.
13 Zhou, Z.; Westover, K. D. Beyond First-Generation KRAS Inhibitors: BBO-8520 Tests the Dual Mechanism Hypothesis. Cancer discovery 2025 , 15 (3), 455-457.
14 Christensen, J.; Hallin, J.; Bowcut, V.; et al. A non-covalent KRASG12D allele specific inhibitor demonstrates potent inhibition of KRAS-dependent signaling and regression of KRASG12D-mutant tumors. 2022.
15 Yadav, V.; Kashif, M.; Kamalia, Z.; et al. Identification of KRAS mutants (G12C, G12D, and G12V) inhibitors. Future medicinal chemistry 2025 , 17 (18), 2221-2234.
16 Zhang, H.; Lin, G.; Jia, S.; et al. Discovery and optimization of thieno3,2-dpyrimidine derivatives as highly selective inhibitors of cyclin-dependent kinase 7. European journal of medicinal chemistry 2024 , 263, 115955.
17 Wang, K.; Zhou, Z.; Ma, X.; et al. Design, synthesis, and bioevaluation of SOS1 PROTACs derived from pyrido2,3-dpyrimidin-7-one-based SOS1 inhibitor. Bioorganic & medicinal chemistry letters 2024 , 107, 129780.
18 Tang, Y.; Pu, X.; Yuan, X.; et al. Targeting KRASG12D mutation in non-small cell lung cancer: molecular mechanisms and therapeutic potential. Cancer gene therapy 2024 , 31 (7), 961-969.
19 Chen, Y. H.; Lv, H.; Shen, N.; et al. EPHA2 feedback activation limits the response to PDEδ inhibition in KRAS-dependent cancer cells. Acta pharmacologica Sinica 2020 , 41 (2), 270-277.
20 van der Hoeven, D.; Cho, K. J.; Ma, X.; et al. Fendiline inhibits K-Ras plasma membrane localization and blocks K-Ras signal transmission. Molecular and cellular biology 2013 , 33 (2), 237-251.
21 Kazi, A.; Xiang, S.; Yang, H.; et al. Dual Farnesyl and Geranylgeranyl Transferase Inhibitor Thwarts Mutant KRAS-Driven Patient-Derived Pancreatic Tumors. Clinical cancer research : an official journal of the American Association for Cancer Research 2019 , 25 (19), 5984-5996.
22 Wang, Y.; Zhong, B.; Xu, C.; et al. Global profiling of AMG510 modified proteins identified tumor suppressor KEAP1 as an off-target. iScience 2023 , 26 (2), 106080.