日, 福建农林大学顾连峰团队在JIPB发表了题为"OmicsCanvas: A multi-omics platform for integration and visualization of epigenetic regulation"的研究论文 (https://doi.org/10.1111/jipb.70258)。该研究针对多组学整合分析中的实际需求, 开发了一个兼具关联分析能力与图形绘制功能的一体化软件流程。

背景与意义
随着高通量测序技术的发展,表观遗传学的研究已经不再局限于单一层面。染色质可及性(ATAC‑seq)、组蛋白修饰(ChIP‑seq)、DNA甲基化(BS‑seq)以及RNA修饰(m⁶A‑seq)等手段能够从不同角度描绘基因调控的图景。但问题也随之而来:这些数据格式不同、分析流程各异,很难放在同一个坐标系里进行比较和关联分析。尤其当研究者想回答"某个基因表达上调,是否伴随启动子区H3K4me3富集和CG甲基化下降"这类问题时,现有的工具要么只能处理一种数据类型,要么做出来的图不够专业、难以直接用于发表。OmicsCanvas提供一个统一的分析和可视化框架,把转录组和多个表观组数据拉到一起看,同时还能生成出版级的矢量图。
方法设计
OmicsCanvas 是一个用 Python 3.9 写的软件包,设计上尽量少依赖外部库,核心依靠 pysam、NumPy、SciPy、scikit‑learn、pandas 和 Matplotlib。它也提供了一个基于 PySide6 (Qt) 的图形界面,可以在 Windows 和 Linux 上跑。
输入层:接受排序并建好索引的 BAM 文件(来自 RNA‑seq、ChIP‑seq、ATAC‑seq、m⁶A‑seq),同时支持 GFF 或 BED 格式的基因注释文件。对于 BS‑seq 数据,它直接读 Bismark 输出的 CX 报告,不需要额外转换。
坐标与分箱:这是比较核心的设计。每个基因区域会被分成基因体(gene body)和两侧的侧翼区(比如上下游各 2 kb),然后把这些区域等距离切成若干个 bin(默认 300 个,用户可以改)。BAM 里覆盖到这些 bin 上的 reads 数会被统计成覆盖度值,还可以选择是否用文库大小做归一化。这样每个基因的每个 bin 就形成了一个统一的行×列信号矩阵,不同组学数据可以在同一个矩阵结构里对齐。
BS‑seq 处理:对 Bismark 的 CX 报告进行解析,在 bin 级别上分别计算 CG、CHG 和 CHH 三种甲基化类型的比例,然后生成元图、热图、单基因图等。
差异表达与转座子:内嵌了一个差异表达模块,对原始的 counts 数据用 PyDESeq2 的中位数比值法来估计大小因子,再拟合负二项回归模型,用 Benjamini‑Hochberg 校正 FDR。如果用户提供了转座子(TE)的 GFF 或 BED 注释,它也能算 TE 的表达量,方便把 TE 相关的组蛋白修饰、染色质可及性和 TE 表达放在一起看。
聚类与富集分析:支持无监督学习(比如 K‑means),可以找出在多条信号轨道上表现相似的基因或元件,输出聚类的热图、空间剖面图和对应的表达分布。同时内置了 GO 富集分析模块,用超几何检验(右侧尾检验)做过表征分析(ORA),再用 BH 法校正 p 值,直接生成气泡图或散点图。
可视化输出:这是 OmicsCanvas 比较出彩的地方。它支持几种出版级图形:
-
元图(metaprofile):展示信号在基因体及两侧的平均分布趋势。
-
热图:单个基因作为一行,按表达量或信号强度排序,观察表观信号和表达的关系。
-
单基因图:针对某一个感兴趣的基因,展示多种组学信号。
-
线性伪 3D 轨迹图:在同一个坐标窗口里堆叠多条轨道,可以分组共享 y 轴范围。
-
Circos 圈图:把同一段基因组区域(比如 ±2 kb)映射到圆周上,基因结构作为内圈,外侧同心圆展示不同组学的覆盖度或甲基化信号。 所有这些图形都可以直接导出为 SVG 或 PDF 矢量格式,不需要再二次修图。
结果展示
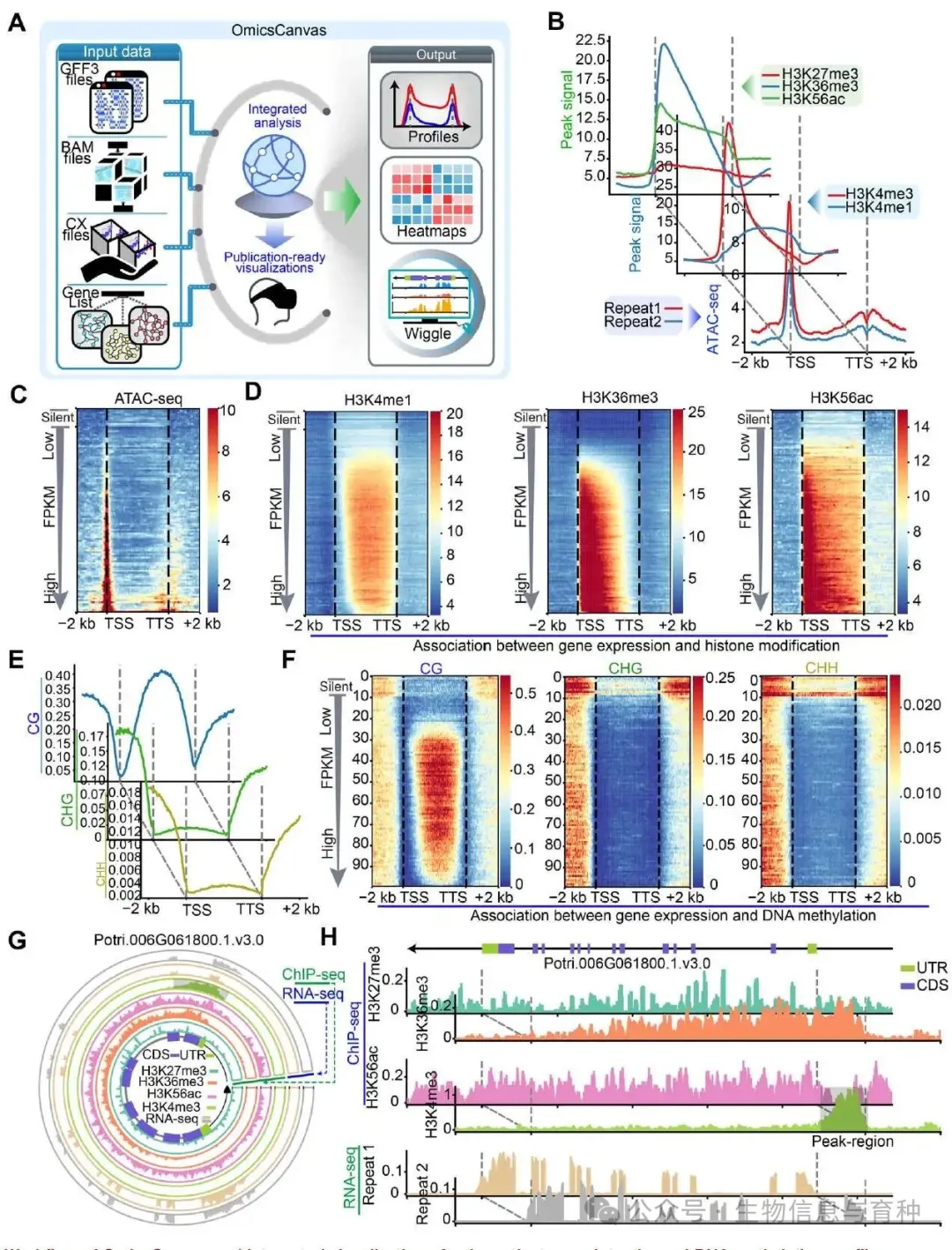
为了演示实际效果,作者用了一个毛果杨(Populus trichocarpa)的数据集,包括 DNA 甲基化和组蛋白修饰数据。
-
组蛋白与染色质可及性的元图:展示了五种组蛋白修饰和 ATAC‑seq 信号在基因体及上下游 ±2 kb 的平均分布。可以看到某些修饰在 TSS 附近富集,有些则在基因体上分布更均匀。
-
表达与表观关联的热图:把基因分成"不表达"和"表达"两组,表达组内再按表达量从低到高排序。在 ATAC‑seq 热图上,表达量高的基因在 TSS 区域明显有更高的染色质可及性。类似地,H3K4me1、H3K36me3 和 H3K56ac 三个组蛋白标记的热图也显示出与表达水平明显相关的富集模式。
-
DNA 甲基化分析:分别对 CG、CHG、CHH 做了元图和热图。元图上可以看到 CG 甲基化在基因体区域有一个典型的"凹谷",而 CHG 和 CHH 整体水平更低且分布不同。热图里把不表达和表达的基因放在一起,能直观看出表达高的基因往往在启动子和基因体上有较低的 CG 甲基化。
-
单窗口圈图:针对一个 ±2 kb 的基因组窗口,从内到外依次绘制了 H3K27me3、H3K36me3、H3K56ac、H3K4me3 和两个重复的 RNA‑seq 信号。不同轨道在同一个圆周上对齐,可以一眼看出某个组蛋白修饰峰和转录信号的位置关系。
-
线性伪 3D 图:在同一段区域里把 H3K27me3、H3K36me3、H3K4me3、H3K56ac 和 RNA‑seq 上下堆叠,并用半透明色块标出了 MACS2 找到的 H3K4me3 峰区域,便于核对 peak 和表达量的对应关系。
此外,文中还提到了可以生成全基因组的 Circos 图(附图 S2)和样本间甲基化相关性的热图(附图 S3),以及聚类后的表达分布图(附图 S4)和 GUI 界面截图(附图 S5)。
讨论与结论
作者认为,OmicsCanvas 的主要贡献不是发明了新的算法,而是把一堆现有的、零散的分析步骤整合进了一个统一的分箱框架里。在这个框架下,RNA‑seq、ChIP‑seq、ATAC‑seq、BS‑seq 和 m⁶A‑seq 的信号可以被放在同样的基因坐标网格中比较,这一点直接解决了多组学分析中数据不齐的痛点。
相比同类工具,它有几个明确的特点:一是既支持基因也支持转座子的定量;二是从差异表达、聚类到 GO 富集可以一条线做下来;三是输出直接就是出版级的矢量图,不需要再用 Illustrator 或 Inkscape 二次加工;四是提供了命令行和图形界面两种使用方式,兼顾服务器批处理和桌面交互。
局限性方面,文中没有刻意回避:它是一个整合型工具,依赖的底层算法(如差异表达、peak calling 等)仍然是调用或借鉴已有方法。但这也正是它的优势------不需要研究者自己写代码去拼接多个软件的输出。
总的来说,OmicsCanvas 降低了对编程能力的要求,让做植物或动物表观组学的人能够比较轻松地把自己的多组学数据变成可解释、可发表的图形。代码和示例数据已经放在 GitHub 上(https:// github.com/zeyubio/OmicsCanvas),遵循最小依赖原则,比较容易安装和运行。
[