请多多关注我的个人网站哦
http://blindbaby.top![]() http://blindbaby.top/
http://blindbaby.top/
我们之前说过,使用GGA或者PBE计算出来的带隙是和实验值有差距的,而Material Project也是用PBE进行计算的,所以这些能直接拿到数据的方式基本都与实际值有一定偏差,那我们要怎么办呢?
https://muchong.com/t-7486590-1![]() https://muchong.com/t-7486590-1
https://muchong.com/t-7486590-1
哎,这时候就要用到HES06或者GW计算了,这两种都是使用的更加精确的计算理论与方法,能得到更加贴近实际值的结果,因为GW之前跑了很久还差点给我电脑跑烧,我们就来讲一讲HES06的流程吧。
HES06与我们讲第五节电子结构计算与光学分析的流程是基本一致的,所以建议你先把第5节看一看
DFT学习记录5电子结构分析+光学分析![]() https://zhuanlan.zhihu.com/p/2017688171392049394
https://zhuanlan.zhihu.com/p/2017688171392049394
我们依然是要进行opt结构优化,然后HES静态计算,拿到CHGCAR与WAVECAR后我们就可以进行下一步了。
首先把POSCAR,POTCAR,KPOINTS,CHGCAR,WAVECAR放到一个文件夹中,接下来运行vaspkit,先像我们在第5节一样生成一下KPATH.in,进入vaspkit进入3
python
301) 1D Structure
302) 2D Structure
303) Bulk Structure
304) K-Path for Wannier90 Code
305) K-Path for Phonopy Code
306) K-Path for CP2K Code
309) Visualize K-Path in First Brillouin Zone
##
常用的就是301 302 303,分别对应三个不同维度的材料,根据你的材料进行选择即可
##生成KPATH.in之后,不要像第5节直接改名为KPOINTS哦,直接运行会报错的,我们要进一步生成HES06专用KPOINTS,打开vaspkit,进入25) Hybrid-DFT Band-Structure
python
================== Hybrid-DFT Band-Structure ====================
250) Generate KPOINTS Including Irreducible Kmesh and Band Edges
251) Generate KPOINTS for Hybrid Band-Structure Calculation
252) Band-Structure for Hybrid-DFT Calculation
253) Projected Band-Structure for Selected One Atom
254) Projected Band-Structure for Each Element
255) Sum of Projected Band-Structure for Selected Atoms
256) Projected Band-Structure by Element-Weights
257) Sum of Projected Band for Selected Atoms and Orbitals
0) Quit
9) Back
------------>>选择251即可生成KPOINTS
最后编写一下INCAR,我让豆包加了一下注释,可以大致参考一下,编写完成后就可以开始运行了,我是R5600+5700xt,跑了大概三四个小时跑完的
python
SYSTEM = XXX2-HSE06-band # 任务名称:能带计算
ISTART = 1 # 读取已有的波函数继续计算
ICHARG = 1 # 读取已收敛的电荷密度文件
ISMEAR = 0 # 半导体/绝缘体用高斯展宽
SIGMA = 0.01 # 展宽系数(小值保证带边准确)
ENCUT = 400 # 平面波截断能
EDIFF = 1E-6 # 能量收敛精度(高精度)
NSW = 0 # 不进行结构弛豫(静态计算)
IBRION = -1 # 关闭离子弛豫
LORBIT = 11 # 输出分波态密度/轨道信息(画图用)
LREAL = .FALSE. # 全空间实空间投影关闭(杂化必选)
NPAR = 4 # 并行参数
PREC = Normal # 精度标准
# ==============================================
# HSE06 杂化泛函标准参数
# ==============================================
LHFCALC = .TRUE. # 开启杂化泛函
HFSCREEN = 0.2 # screened 交换范围
AEXX = 0.25 # HF交换占比 25%
AGGAC = 0.75 # GGA相关占比 75%
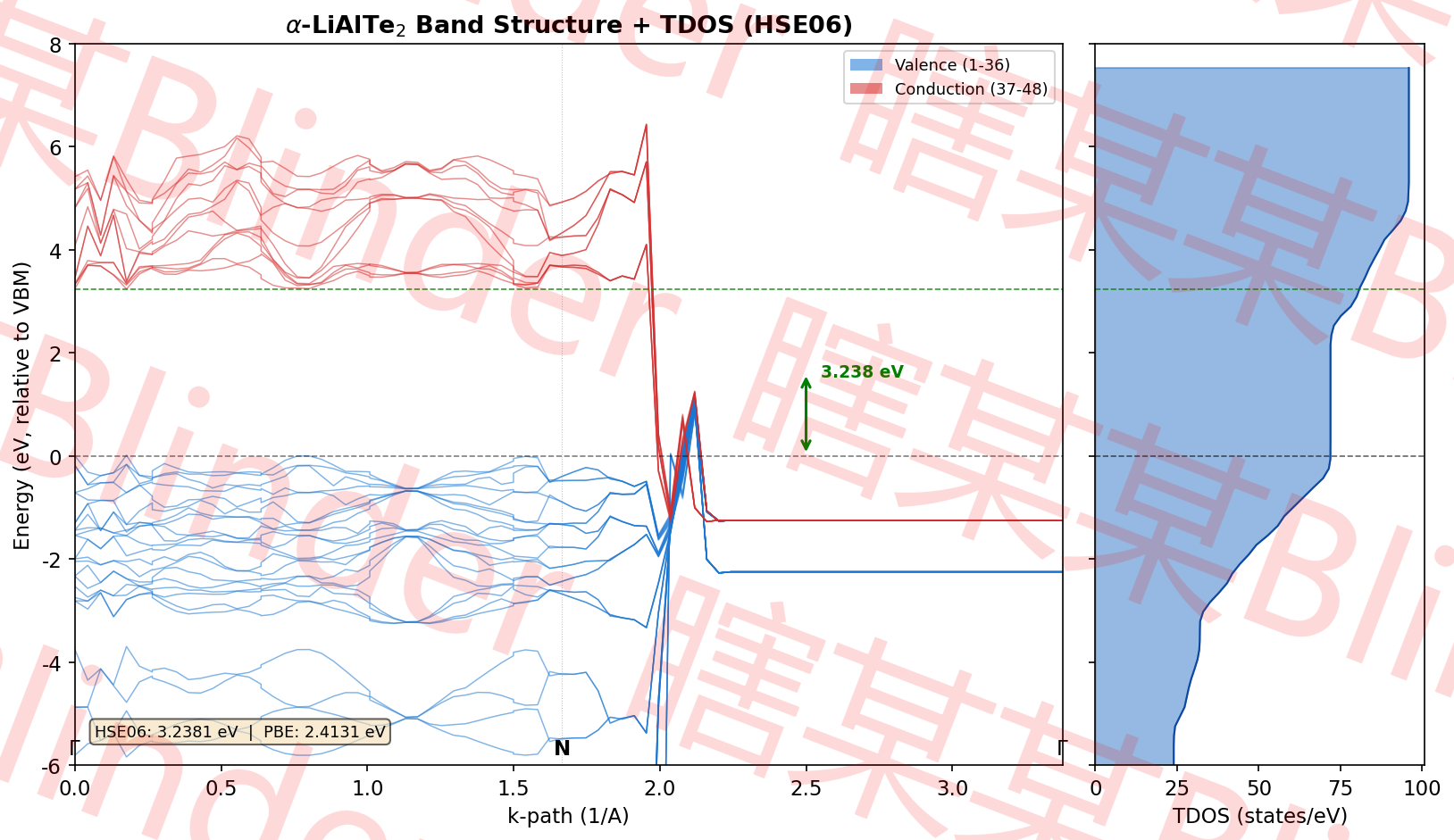
PRECFOCK = Fast # 快速杂化计算(提速)计算完成之后使用vaspkit的25中的 252) Band-Structure for Hybrid-DFT Calculation 即可查看计算完成后的能带图与.dat,这是我用.dat数据画出来的图(请勿直接用,如果你需要帮助可以在我的个人网站看到我的邮箱和我联系,或者直接私信)
至于空穴有效质量,只需要在这个基础上继续进行我们之前第4节的内容就够了
DFT学习记录4 电子和空穴的有效质量计算![]() https://zhuanlan.zhihu.com/p/2005427782205843336
https://zhuanlan.zhihu.com/p/2005427782205843336
如果你要做DOS态密度分析的话,那基本和PBE计算没有区别,就是vaspkit多跑一下,还是继续去看第5节即可。
我在对LiAlTe2进行计算之后,发现PBE算出来的带隙宽度为2.41eV,material project里的为2.44eV,使用HES06计算出来的为3.24eV,这就和历史研究中的结果非常接近了。
对于空穴有效质量,不管是用PBE还是用HES06,结果其实都差不多,都在0.68左右,这么来看的话,泛函选择对空穴有效质量的影响还是不大的,但是对于电子有效质量,则有比较大的偏差。
光学分析的话,我个人感觉没太大必要,因为HES06计算光学的时间会比能带时间长特别特别多,不如直接PBE算然后用修正后的能带去做修正