YaHS: yet another Hi-C scaffolding tool
YaHS:一款新型 Hi-C 挂载组装软件
概述
本研究介绍YaHS :一款操作简便的命令行工具 ,可基于 Hi-C 数据构建染色体水平基因组支架 。该工具仅需单行命令 即可运行,对用户输入要求极低(仅需组装文件与比对文件),输入格式兼容同类软件,并支持多种输出格式;能够快速、稳定、可扩展 地完成高质量基因组组装,兼具高准确性与高连续性。
可用性与实现
YaHS 采用C 语言 开发,基于MIT 开源协议 授权。源代码、使用文档及教程可访问:https://github.com/sanger-tol/yahs。
1 引言
长读长单分子测序技术在读长、碱基准确率与单位碱基测序成本上的飞速革新,开启了从头基因组组装的黄金时代。近几年多项大型基因组测序项目陆续启动,包括地球生物基因组计划、脊椎动物基因组计划与达尔文生命之树计划,旨在为数千种基因组大小、复杂度与倍性各不相同的物种组装高质量染色体水平基因组。尽管测序技术不断进步,仅依靠长读长测序仍难以获得参考级基因组。
研究者常借助物理图谱、遗传图谱、光学图谱、Hi-C 互作图谱等长距离连锁信息,将重叠群(contig)锚定拼接为染色体水平支架。Hi-C 是基于邻近连接的测序技术,可获得基因组位点间的互作信息,最初用于研究细胞核内基因组三维空间结构。由于位点间互作频次与基因组上的物理距离高度相关,Hi-C 凭借性价比优势,迅速成为构建染色体水平基因组支架的主流手段。
目前已开发多款基于 Hi-C 的挂载软件,包括 LACHESIS、HiRise、3D-DNA、ALLHiC、SALSA2、pin_hic 等,但各类工具均存在局限性,组装效果受基因组复杂度、重复序列占比、Hi-C 文库构建质量、测序深度等多种因素影响。
本文介绍YaHS ,一款依托 Hi-C 数据实现染色体水平支架构建的新型组装工具。YaHS 遵循 Hi-C 挂载通用流程:Hi-C 测序读段比对至初始重叠群、按需拆分错误重叠群、构建互作矩阵、搭建并精简组装图、最终输出支架序列;软件默认输入重叠群源自单一单倍型,若基因组组装结果含有单倍型冗余序列,需提前剔除。
YaHS 区别于同类软件的核心创新在于全新的互作矩阵构建算法 ,能够更精准判断重叠群的连接关系。在模拟数据与真实数据集上,同 SALSA2、pin_hic 两款主流软件对比测试表明:YaHS 组装结果的准确度、连续性更优,对原始组装错误的容错能力更强。
2 结果
2.1 软件整体流程概述
Hi-C 测序读段与重叠群的序列比对不在 YaHS 程序范围内。本研究采用 Arima 官方比对流程,包含四步:单端模式序列比对、跨连接嵌合序列过滤、读段配对、PCR 重复去除。
YaHS 仅需 BAM 或 BED 格式的比对文件作为输入,可选择性在无 Hi-C 覆盖的疑似错误位点拆分重叠群,随后进行多轮迭代挂载组装:每一轮组装中,程序将所有重叠群切分为固定长度的分块(即分辨率),把 Hi-C 互作信号统计至分块对应的矩阵单元格;同一条重叠群内部单元格为胞内单元,不同重叠群间单元格为胞间单元 ,逐一统计单元格互作频次。
计算两重叠群连接得分时,利用同间距胞内单元的中位数作为期望基线,对胞间互作频次做归一化,再加权求和得到连接分值。核心逻辑:物理相邻的重叠群,其跨片段胞间互作频次应与同间距胞内互作频次水平接近。程序会计算两个重叠群四种排布方向的连接分值,择优确定最终连接朝向;若导入 Hi-C 所用限制性内切酶信息,软件可先依据酶切位点数量校正单元格互作频次。
以重叠群为节点、连接关系(加权得分)为边构建组装图,依次执行低分边过滤、短枝修剪、平端修整、重复区拆解、传递边删除、环状气泡消除、朝向歧义修正、弱边与可疑连接剔除等步骤简化网络图;沿连续路径遍历图谱,生成基因组支架。程序可选二次纠错:在缺乏有效 Hi-C 互作支撑的连接处拆分支架。
YaHS 采用多轮层级组装策略 ,随组装轮次提升逐步降低分辨率、增大分块长度;除首轮直接使用原始 contig 外,后续每轮均以上一轮组装得到的支架作为输入,详细原理见补充材料。
2.2 基于人类基因组模拟数据集测试
将端粒到端粒完整人类参考基因组 T2T-CHM13 随机打断为 100 kb~1 Mb 片段,得到含 5483 条 contig、N50=715 kb 的模拟组装数据集;从 NCBI 下载配套 Arima-Hi-C 测序数据,分别使用 YaHS、SALSA2、pin_hic 开展挂载。
YaHS 可将 92% 以上序列组装至 25 条大于 35 Mb 的主支架,组装 N50=132.6 Mb(L50=9)、N90=36.5 Mb(L90=23);SALSA2 组装 N50=43.9 Mb(L50=22)、N90=3.3 Mb(L90=103);pin_hic 组装 N50=51.5 Mb(L50=19)、N90=8.5 Mb(L90=68)。
利用 QUAST-LG 软件将组装支架比对至 T2T 参考基因组评估错误,组装错误分为三类:片段移位、片段倒位、染色体间异常易位。YaHS:移位 40 处、倒位 21 处;SALSA2:移位 262 处、倒位 55 处;pin_hic:移位 171 处、倒位 115 处;三款软件均未检出染色体间易位错误。
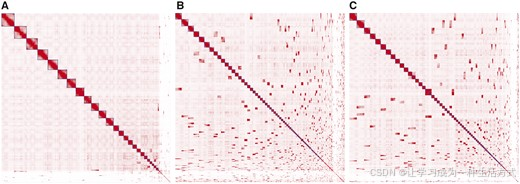
图 1 无组装错误的 T2T 模拟数据集分别经 YaHS (A)、SALSA2 (B)、pin_hic (C) 挂载后基因组 Hi-C 互作热图
颜色深浅代表横、纵坐标对应基因组位置间 Hi-C 测序配对 reads 的富集密度,像素颜色越深代表互作信号越高;主对角线上方框标注区块为软件拼接得到的基因组支架;对角线外深色区块代表可进一步拼接、合并为更大支架的支架组合。热图由 Juicebox 软件绘制 (Durand 等人,2016)。
引入组装错误的软件性能测试
为测试三款软件对错误组装的纠错能力,在上述模拟组装序列中人工插入 25 条错误重叠群,合计 30 处组装错误:10 处为同染色体两段序列错误连接、10 处为跨染色体序列错误拼接、5 处为三段序列两两错连,随机设定片段连接方向,错误序列总长 32.1 Mb。修正后数据集共 5453 条 contig,N50=718 kb。
- YaHS 成功校正 30 处错误中的 28 处;剩余未修正错误来自 1 条双重错配片段:19 号染色体短片段(170 kb)两侧分别衔接两段间距 1.2 Mb 的 10 号染色体序列,该构型纠错难度高(附图 S1);YaHS 与 SALSA2 均未出现误切 contig 的假阳性纠错。
- SALSA2 仅校正 30 处错误中的 14 处。
- pin_hic 无独立纠错步骤,仅在挂载末尾对可疑错误位点拆分支架。
纠错后组装连续性:YaHS 各项组装指标与原始无错组装完全一致;SALSA2 组装 N50 小幅升至 46.1 Mb、L50 降至 20;pin_hic 组装碎片化加重,N50 降至 37.5 Mb (L50=28)、N90 降至 5.8 Mb (L90=98)。借助 QUAST-LG 比对参考基因组统计组装缺陷:受未修正错误片段影响,YaHS 仅新增 2 处移位、2 处易位错误;SALSA2 组装出现移位 278 处、倒位 18 处、易位 63 处;pin_hic 组装移位 188 处、倒位 22 处、易位 123 处。
2.3 真实人类基因组组装测试
采用 PacBio 测序得到的 CHM13 原始 contig 组装序列 (NCBI 登录号:GCA_000983455.2) 开展实测:该组装含 4961 条 contig,总长度 2.94 Gb,N50=10.5 Mb (L50=82)、N90=972 kb (L90=403),沿用前文同一套 Hi-C 数据。挂载时 YaHS 拆分 125 处错误 contig,SALSA2 仅拆分 20 处;三款软件组装结果:
- YaHS:N50=147.7 Mb(L50=8),N90=39.4 Mb(L90=22)
- SALSA2:N50=102.7 Mb(L50=11),N90=8.1 Mb(L90=42)
- pin_hic:N50=87.9 Mb(L50=12),N90=14.6 Mb(L90=38)
比对 T2T-CHM13 参考基因组,QUAST-LG 统计总组装错误依次为 2627、2835、2799 个;绝大多数错误源自前期 PacBio 组装缺陷,对应数值分别为 2127、2260、2258 个。得益于更强的纠错能力,YaHS 修正的原始组装错误最多。支架挂载衍生错误(移位 / 倒位 / 易位):YaHS:442/12/46;SALSA2:468/16/91;pin_hic:446/6/89。三款组装对应的 Hi-C 互作热图见附图 S2。
2.4 达尔文生命之树 (DToL) 项目物种批量测试
选取 DToL 项目中 15 个分属不同类群、基因组大小与原始组装质量各异的物种,利用三款软件分别挂载。综合 N90 等组装连续性指标,YaHS 组装结果整体优于 SALSA2、pin_hic (附表 S1,附图 S3~S17)。
以 ** 丛蟋 (Meconema thalassinum,物种编号 iqMecThal1)** 为例:预估基因组超 9 Gb,原始组装共 2093 条 contig,总长 9054 Mb,N50=10.7 Mb (L50=229)。YaHS 识别 268 处组装错误;最终组装中 3 条长度>1349 Mb 的支架占基因组总长 50%,13 条>179.6 Mb 的支架覆盖全基因组 90%,最大支架长度达 2087 Mb。对照:SALSA2 组装 N50=79.0 Mb (L50=18)、N90=3.5 Mb (L90=311);pin_hic 组装 N50=202.4 Mb (L50=12)、N90=14.2 Mb (L90=82),组装热图见附图 S3。另一个物种硫黄菌 (Laetiporus sulphureus,编号 gfLaeSulp1) 测试中,YaHS 与 pin_hic 均在端粒区域出现少量错误拼接(附图 S17)。
3 结论
YaHS 是一款依托 Hi-C 数据构建染色体水平基因组支架的高速、稳定、高精度工具,现已在达尔文生命之树等科研项目中常规投入使用。在跨度广泛的物种、基因组尺寸与初始组装质量条件下,YaHS 的组装准确度与连续性整体优于现有主流 Hi-C 挂载软件;该软件可依托实测数据自主优化参数,对不同建库方案、不同基因组距离分布特征的 Hi-C 数据兼容性强。软件开源免费、使用便捷、配套文档完善。