Pangenome analysis provides insights into legume evolution and breeding
泛基因组解析助力阐明豆科植物演化规律并为育种研究提供理论依据

摘要
豆类粮食作物是发展可持续农业的优质种质资源。目前学界虽已开展大量豆科物种进化研究,但调控适应性演化、支撑农艺性状改良的保守分子机制仍尚不明确。本研究完成菜豆、鹰嘴豆、豌豆、小扁豆、蚕豆、木豆、豇豆、绿豆、荷包豆 9 种主食用豆类的高质量基因组组装。泛基因组分析表明,冷季型与暖季型豆类各自发生特异性基因家族扩张,新基因诞生与基因复制在根瘤结瘤自主调控通路演化中发挥关键作用。研究发现,数百个基因在豆科进化历程中受到趋同选择,调控粒重等重要农艺性状。此外,冷季豆类基因稀疏区中转座子的串联扩增,是推动基因组扩张、形成顺式调控元件的重要驱动力。上述研究结果解析了豆科物种分化的分子基础,同时为豆类遗传育种提供宝贵基因组资源。
正文
豆科(Fabaceae,又称 Leguminosae)是被子植物中物种数量排名第三的大科。豆类作物可为人类提供优质食用蛋白,还能通过生物固氮提升土壤肥力,是支撑可持续农业发展、保障全球粮食安全不可或缺的重要种质资源。依据适宜生长的气候时节,食用豆类可划分为冷季型与暖季型两大类。鹰嘴豆、豌豆、小扁豆、蚕豆、白羽扇豆等冷季豆类在晚春至初夏完成生育周期;而大豆、菜豆、木豆、豇豆、绿豆、藊豆等暖季豆类,最适生长期集中在晚春至初秋的高温时段。
约 6500 万年前,豆科祖先物种共同经历一次全基因组复制(WGD)事件;后续部分豆科支系又发生了新近多倍化,例如大豆属物种在约 1300 万年前发生一次特有的全基因组复制。与此同时,转座子(TE)扩张重塑了豆科基因组结构,造成豆科基因组大小差异悬殊,基因组跨度在 0.4~13 Gb 之间。食用豆类与禾本科谷物在全球多地同步被驯化,随后逐步引种扩散至各类生态地理区。尽管各豆类物种演化相互独立,但长期人工选择趋同塑造了作物性状:普遍表现为种子休眠性减弱、籽粒变大。
本研究对 9 种豆类开展从头基因组组装,并结合泛基因组解析,挖掘豆类环境适应性进化与人工驯化背后的遗传变异规律。研究筛选到数百个在多物种驯化过程中遗传多样性显著下降的基因,同时提出转座子串联扩增模型,用以解释冷季豆类基因组膨大的分子成因。上述结果为解析豆科演化机制提供新见解,也为豆类种质改良提供优质基因组数据资源。
结果
食用豆类从头基因组组装
为解析豆科遗传多样性与适应性进化规律,本研究选取全球主栽的 9 种食用豆类开展高质量从头组装,包含菜豆(苏菜豆 19-17)、鹰嘴豆(XJ01)、豌豆(苏豌豆 08)、小扁豆(SX01)、蚕豆(P17-186)、木豆(SM01)、豇豆(苏豇豆 01)、绿豆(苏绿豆 07)、藊豆(边红 01,图 1a、1b)。上述物种虽已有基因组报道,但菜豆、木豆、鹰嘴豆等早期版本均基于短读长测序搭建,组装质量有待优化。本研究累计获得 732.7 Gb PacBio HiFi 测序数据,各物种测序深度 20~50 倍(附表 1),依托 HiFi 数据完成初级 contig 组装;针对鹰嘴豆额外补充 42.8 Gb 牛津纳米孔超长读长数据,单独组装得到 contig 后与 HiFi 拼接序列整合优化。借助 Hi-C 染色质构象捕获数据(测序深度 80~140 倍,附表 1),全部物种均提升至染色体水平组装,基因组大小区间 463.3 Mb~13.0 Gb(扩展数据图 1、附表 2)。共线性比对显示,本版基因组与已发表参考基因组共线性保守度高(扩展数据图 2a--h),整体组装质量优于现有公开版本。以鹰嘴豆为例:本研究组装基因组总长约 670 Mb,仅 69 条 scaffold,染色体挂载率超 99.6%;而目前应用广泛的 CDC Frontier 参考基因组总长仅 530 Mb,由数千条 scaffold 构成。
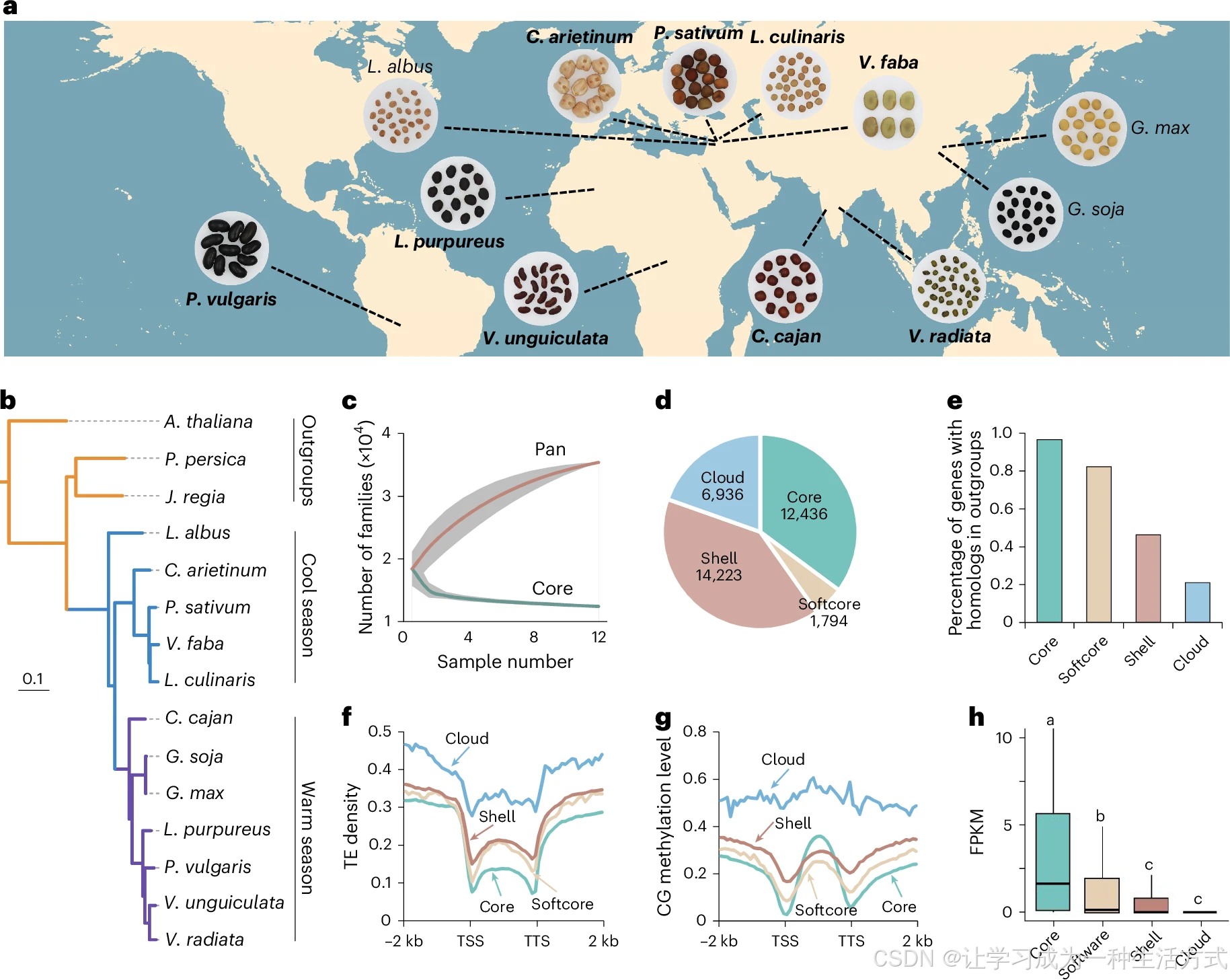
a. 豆类作物地理分布图;加粗物种为本研究完成基因组组装材料,包括菜豆(Phaseolus vulgaris ,苏菜豆 19-17)、鹰嘴豆(Cicer arietinum ,XJ01)、豌豆(Pisum sativum ,苏豌豆 08)、小扁豆(Lens culinaris ,SX01)、蚕豆(Vicia faba ,P17-186)、木豆(Cajanus cajan ,SM01)、豇豆(Vigna unguiculata ,苏豇豆 01)、绿豆(Vigna radiata ,苏绿豆 07)、藊豆(Lablab purpureus ,边红 01)。b. 基于最大似然法构建的豆科系统发育树,拟南芥、桃、核桃作为外类群。c. 随基因组数量增加,泛基因家族与核心基因家族的数量变化,数据以平均值 ± 标准差表示。d. 核心基因、软核心基因、壳基因与独有基因家族的占比。e. 四大类基因中存在外类群同源基因的基因占比。f. 转座子(TE)在核心、软核心、壳、独有基因上下游的分布特征。g. CG 型甲基化在四类基因上下游的分布模式。h. 核心基因(30891 个)、软核心基因(3806 个)、壳基因(15511 个)、独有基因(572 个)的基因表达量。柱上方不同小写字母代表组间在\(P<0.05\)水平差异显著;显著性由单因素方差分析(ANOVA)结合 Scheffe 多重比较检验。箱线箱体上下边界分别为 75%、25% 分位数,中线为中位数,须线延伸至 1.5 倍四分位距(IQR)。TSS:转录起始位点;TTS:转录终止位点。
各基因组中转座子(TE)占比介于 51%~92%(附表 3)。本研究在 9 种豆类基因组中注释得到 26180~55519 个基因模型,绝大多数基因组的 BUSCO 完整基因检出率高于 99%(附表 2)。相较于已发表的基因注释版本,新组装基因组新增 727~27995 个基因,其中超 88% 的基因编码蛋白含有 Pfam 数据库收录的保守结构域(扩展数据图 2i)。结合本研究 9 个豆类基因组、栽培大豆、野生大豆与白羽扇豆基因组数据开展泛基因组分析,全部基因聚类得到 35389 个基因家族。随参考基因组数量不断增加,基因家族总数逐步上升,而核心基因家族数量持续下降,与泛基因组变化规律相符(图 1c)。依据在 12 个基因组中的出现频次,将基因家族划分为四类:在全部 12 个物种中均存在的 12436 个家族为核心基因家族 ;在 10 或 11 个物种中出现的 1794 个家族为软核心基因家族 ;分布于 2~9 个物种的 14223 个家族为壳基因家族 ;仅单一物种特有的 6936 个家族为独有(cloud)基因家族 (图 1d)。核心基因家族的基因数量占各物种总基因数的 41%~70%(扩展数据图 3a)。
与核心基因相比,非必需基因(软核心、壳、独有基因)功能保守性更低,在拟南芥、桃、核桃外类群中直系同源基因占比更少(图 1e);其Ka/Ks 比值更高、含 Pfam 结构域的注释比例更低(扩展数据图 3b、3c)。此外,非必需基因的基因区内含更多转座子插入,多富集在异染色质区域(图 1f、扩展数据图 3d),伴随更高水平 DNA 甲基化修饰(图 1g、扩展数据图 3e、3f),染色质开放程度更低、H3K4me3 与 H3K27ac 等活化组蛋白修饰富集度偏弱(扩展数据图 3g~i),最终整体基因表达量显著低于核心基因(图 1h)。独有基因整体表达偏低,但仍有约 65% 的独有基因至少在一个组织中检测到表达(FPKM>0,扩展数据图 3j)。GO 富集结果显示:核心基因显著富集于含氮化合物生物合成、RNA 生物合成等基础生命过程;非必需基因则富集在逆境应答等生物学通路(扩展数据图 3k、3l)。
结瘤相关基因的演化规律
植物与根瘤菌形成的根瘤共生(RNS)呈间断式分布于豆目、蔷薇目、壳斗目、葫芦目 4 个目,共同构成固氮分支(NFC)。根瘤共生的起源与演化存在两种主流假说:一是祖先物种单次演化获得结瘤能力,后续多数支系陆续丢失该性状;二是不同物种独立平行演化形成结瘤特性。本研究依托大豆已发表转录组数据筛选得到 1305 个根瘤偏好表达基因(附表 4),其中约 54% 属于核心基因,这类基因显著富集在嘌呤合成、物质转运、细胞稳态调控、细胞分裂素应答与转录因子功能;剩余非必需基因主要富集在细胞刺激应答、信号传导与金属离子结合相关功能(扩展数据图 4a)。依据系统发育关系将大豆结瘤相关基因划分为 0~7 共 8 个保守等级,0 级跨物种保守度最低,7 级保守度最高(图 2b、方法部分)。结果显示,超 70% 的根瘤偏好基因在固氮分支以外的植物类群中同样存在(图 2c);已被功能克隆的结瘤关键基因中 81% 以上为核心基因,且 90% 可在非固氮分支物种中找到同源序列(扩展数据图 4b、4c)。现有研究表明,根瘤共生改造利用了古老丛枝菌根(AM)共生的关键功能元件。本研究比对发现:结瘤因子识别(NFP、LYK3 )、钙信号通路(DMI1/DMI2/DMI3、IPD3 )、结瘤特异转录因子(NSP1、NSP2 )等固氮通路核心基因,在被子植物、裸子植物、角苔、地钱中普遍保守,而上述物种均可与丛枝菌根真菌建立共生关系(扩展数据图 4d)。此外,超过 55% 的根瘤偏好基因、77% 已克隆结瘤基因在裸子植物、角苔或地钱中存在保守同源基因(扩展数据图 4e)。
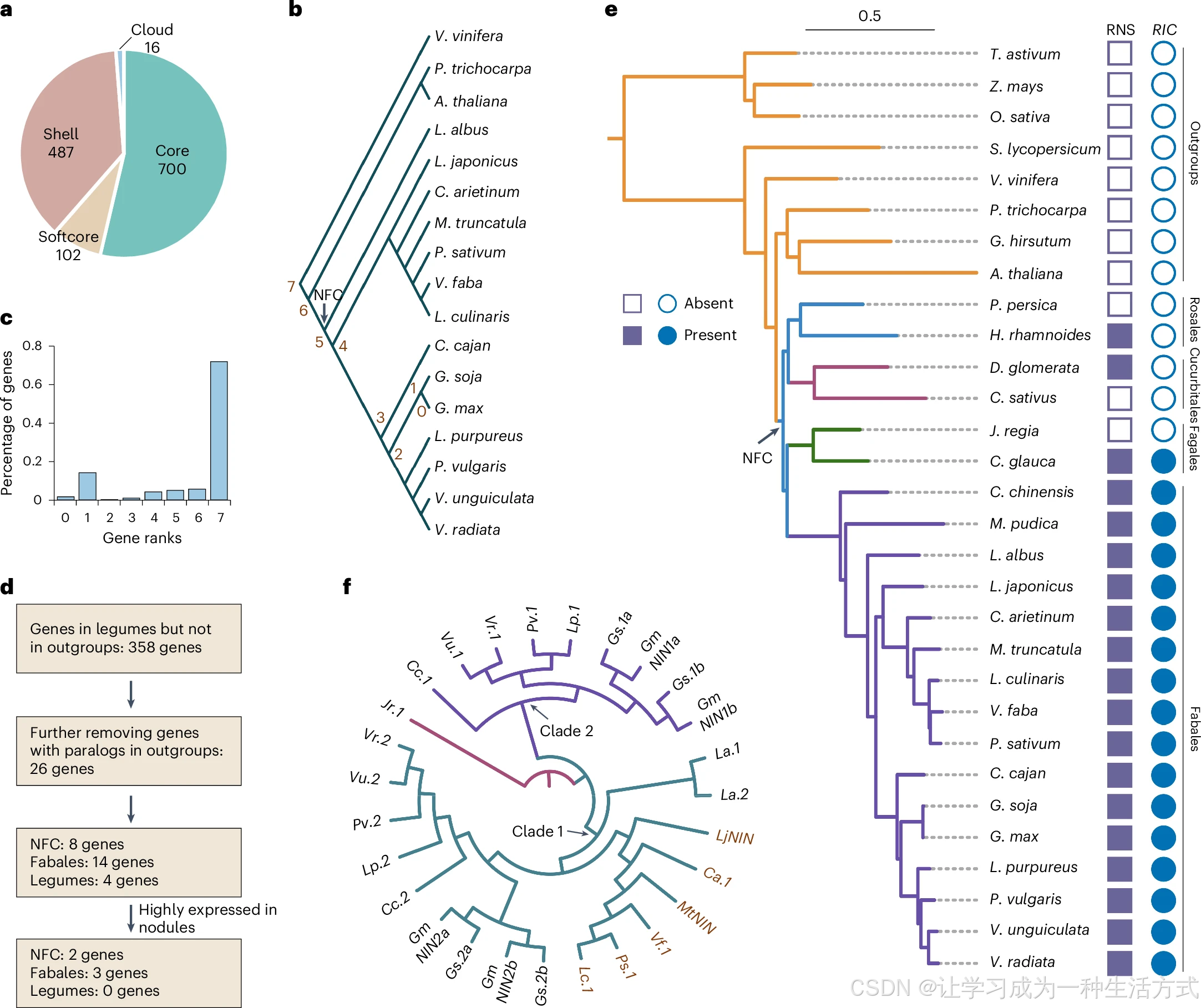
a. 根瘤偏好表达基因中核心、软核心、壳、独有基因的占比分布。b. 标注基因保守等级的豆科系统发育树;大豆基因被划分为 0~7 共 8 个等级,0 级代表大豆物种特有基因 。c. 不同保守等级中根瘤偏好基因的占比。d. 豆科从头起源新基因(de novo 基因)筛选技术路线。e. RIC 基因家族系统发育分析。f. NIN 基因系统发育树。
物种缩写注释: La:白羽扇豆;Vf:蚕豆;Lc:小扁豆;Ps:豌豆;Ca:鹰嘴豆;Mt:蒺藜苜蓿;Lj:百脉根;Cc:木豆;Lp:藊豆;Pv:菜豆;Vu:豇豆;Vr:绿豆;Gm:栽培大豆;Gs:野生大豆;Jr:核桃。
新生从头起源基因(de novo 基因)已在诸多物种中被报道,但该类基因在固氮共生演化中的功能尚不明确。本研究以玉米、水稻、小麦、番茄、葡萄、毛果杨、拟南芥、陆地棉、核桃、黄瓜、桃共 11 个物种作为外类群开展比较基因组分析(扩展数据图 4f),筛选得到 358 个豆科特有、外类群缺失 的基因(图 2d);进一步剔除与外类群基因同源性较高(E 值<\(10^{-10}\))的基因后,最终保留 26 个候选新生基因用于后续分析(图 2d、附表 5)。依据演化节点划分,其中 8 个基因大概率在固氮分支演化阶段新生,14 个起源于豆目分化时期,剩余 4 个在豆科共同祖先阶段产生(图 2d、附表 5)。其中 5 个基因在根瘤中优势高表达,大豆GmRIC2 位列其一,该基因编码受根瘤菌诱导的 CLE 多肽(图 2e)。大豆GmRIC1/GmRIC2 被激活后,通过结瘤自主调控(AON)负反馈通路抑制根瘤过度发生。上述结果表明,演化出固氮性状的豆科植物借助新生基因完善长距离负反馈调控网络,以此平衡植株碳源与氮素供给。
结瘤起始基因NIN 是调控根瘤菌侵染与根瘤器官建成的核心转录因子。固氮分支内部分物种丧失结瘤能力,多与NIN 基因多次独立丢失或片段化相关。NIN 系统发育树将豆类基因分为两大分支(图 2f):冷季豆类的全部 NIN 基因仅聚在分支 1 ;而暖季豆类携带 2~4 个NIN 拷贝,基因分散分布于分支 1 与分支 2。推测暖季豆类NIN 基因发生复制扩增是物种对生存环境变化的适应性演化结果。
豆科演化过程中的基因家族扩张与收缩
冷季豆类与暖季豆类约在 5500 万年前发生分化(扩展数据图 5a)。以白羽扇豆为外类群,借助 CAFE5 软件筛选分化后显著扩张的基因家族(\(P<0.05\)):冷季豆类、暖季豆类分别鉴定到 40 个、37 个显著扩张家族,两类豆类仅有 1 个共同扩张的基因家族(图 3a、扩展数据图 5a、附表 6)。GO 富集显示:冷季豆类特化扩张基因集中在碳固定、光合作用、氧化还原、ATP 代谢及类囊体膜相关通路(图 3b);暖季豆类特异扩张基因主要参与抗病防御、蛋白激酶功能以及逆境、激素应答过程(图 3c)。调控低温适应性的CBF 抗寒基因家族在冷季豆类中显著扩张(扩展数据图 5b);NLR 抗病蛋白是植物抵御病原菌侵染的关键基因,高温环境易促使病原增殖并抑制植物免疫,统计显示暖季豆类 NLR 基因数量显著高于冷季豆类 (图 3d)。上述结果证实,冷、暖季豆类依靠差异化的基因家族演化,分别适应各自独特的生态环境。
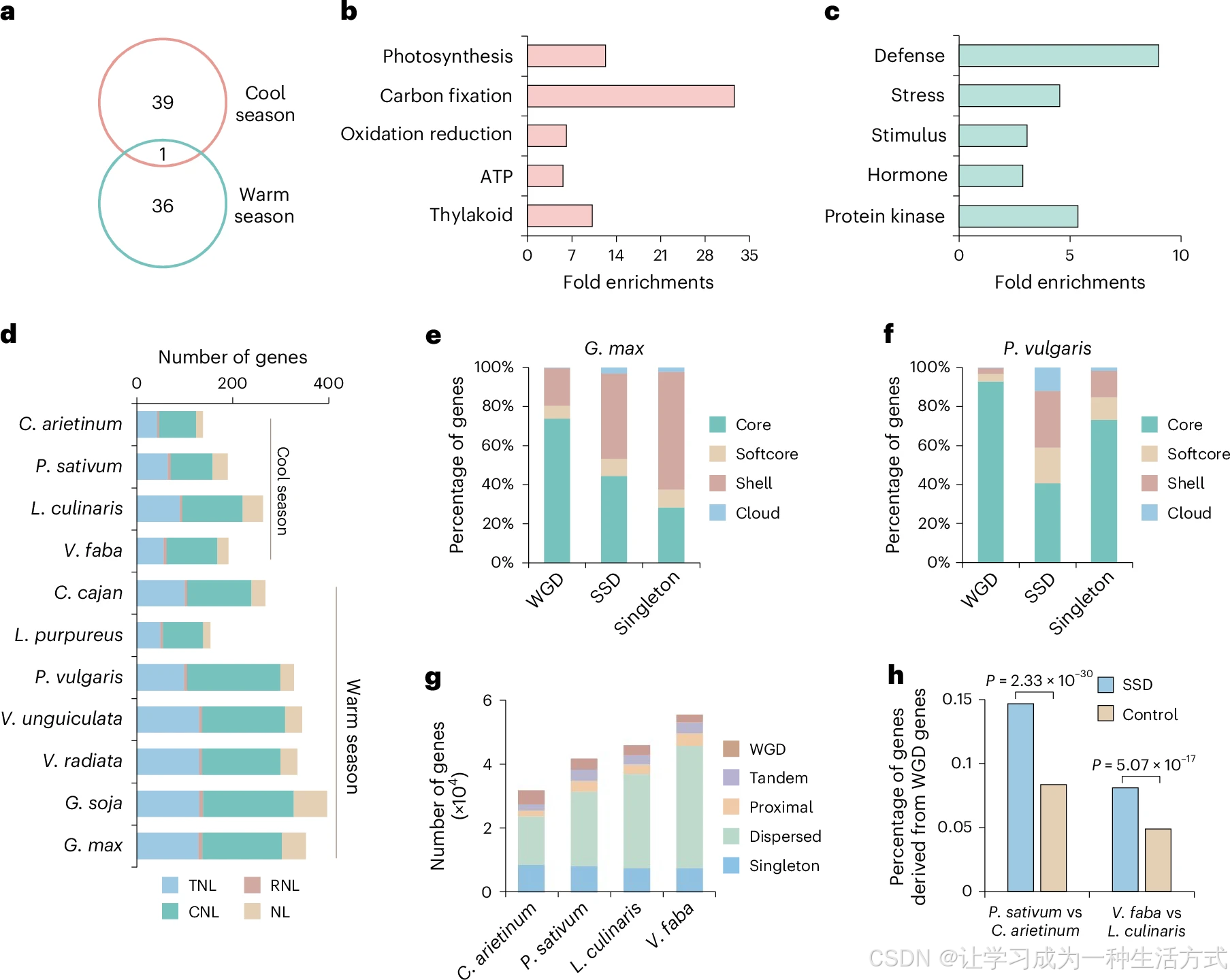
a. 韦恩图展示冷季、暖季豆类扩张基因家族的重叠情况。b、c. 冷季豆类(b)与暖季豆类(c)扩张基因家族富集的显著 GO 条目。d. 不同豆科物种间 NLR 抗病基因拷贝数对比。e、f. 大豆(e)、菜豆(f)中全基因组复制基因(WGD)、小规模复制基因(SSD)、单拷贝基因(singleton)里核心、软核心、壳、物种特有基因的占比。g. 鹰嘴豆、豌豆、小扁豆、蚕豆的 WGD 基因、SSD 基因(串联、近缘、散在复制)与单拷贝基因数量统计。h. 豌豆相对鹰嘴豆、蚕豆相对小扁豆,由古老 WGD 基因新生形成的 SSD 基因占比;基因组其余基因为对照组。显著性采用双侧费希尔精确检验。缩写释义 TNL:含 TIR 结构域的 NLR 基因;CNL:同时含 CC 与 NB-ARC 结构域的 NLR 基因;RNL:含 RPW8 结构域的 CCR-NLR 基因;NL:仅含 NB 与 LRR 结构域的 NLR 基因。
多倍体演化通常始于全基因组复制(WGD)事件,随后经历基因组重排与二倍化进程中的大量基因丢失。古多倍体物种的基因可划分为 WGD 来源基因、小规模复制(SSD)基因和单拷贝基因三类。大豆属物种共鉴定出 31900~33700 个大豆特有 WGD 基因;其余暖季豆类仅保留 2800~3500 个豆科祖先 WGD 衍生基因。无论豆科祖先 WGD 基因还是大豆属特有 WGD 基因,非必需基因占比均不足 30%(图 3e、3f,扩展数据图 5c--f),但大豆属中单拷贝基因里约 70% 为非必需基因(图 3e,扩展数据图 5c)。以上结果说明:二倍化过程中非必需基因更易发生成对拷贝中的单拷贝丢失。
仅经历约 6500 万年前豆科共同 WGD 的暖季豆类中,SSD 基因内软核心、壳基因与物种特有基因占比,显著高于 WGD 基因和单拷贝基因(图 3f,扩展数据图 5d--f)。鉴于非必需基因在逆境应答中发挥关键作用,SSD 中非必需基因富集可弥补二倍化造成的基因丢失,保障作物在多变逆境环境下正常生长。冷季豆类未经历近期全基因组复制,但基因总数从鹰嘴豆 31834 个上升至蚕豆 55519 个,增幅约 1.7 倍(图 3g),基因数量扩张主要由 SSD 复制驱动 。相较鹰嘴豆,豌豆新生 SSD 基因 3283 个;相较小扁豆,蚕豆新生 SSD 基因 4048 个;比较分析证实,近期新生 SSD 基因显著偏好起源于远古 WGD 留存基因(图 3h)。
豆科演化中的趋同选择
保守基因受平行选择是不同物种趋同演化出相似农艺性状的重要成因,但豆科中调控重要性状的基因是否普遍经历趋同选择、选择强度如何仍缺乏系统解析。本研究依托大豆、木豆、鹰嘴豆、豌豆地方种质与野生资源重测序数据集,挖掘距今 3000 万~5500 万年独立分化进程中跨物种趋同受选择位点(扩展数据图 5a)。以群体遗传多样性降幅前 90% 作为受选择阈值:大豆筛选 395 个受选择区段(7278 个基因)、木豆 595 个区段(9224 个基因)、鹰嘴豆 470 个区段(9970 个基因)、豌豆 5407 个区段(14799 个基因)(图 4a,附表 7)。其中 226 个基因在四大豆类中同步出现遗传多样性衰减(图 4b、4c,附表 8),功能富集于种子休眠、籽粒膨大、能量稳态等通路。
大豆GmYUC4a 为生长素合成 YUCCA 家族成员,其直系同源基因木豆CcYUC4 、鹰嘴豆CaYUC4 、豌豆PsYUC4 在各自物种驯化中同步发生遗传多样性下降(图 4d,扩展数据图 6a、6b、图 7,附表 8);拟南芥同源基因YUC1/YUC4 通过提升内源生长素积累正向调控籽粒大小。依托大豆公共组学数据库 SoyOmics,依据 3'UTR 插入缺失与下游 SNP 标记将GmYUC4a 划分为 Hap1~Hap3 三种单倍型(图 4e);携带 Hap1 的大豆籽粒重量显著低于 Hap2、Hap3 型(图 4f)。群体基因型统计:野生大豆 Hap1 占比>94.5%,地方品种降至 4.3%,现代栽培种仅 0.4%(扩展数据图 6c--e)。随机选取含 Hap1、Hap2 的大豆材料进行表达检测,GmYUC4a 在 Hap2 株系中表达量更高(扩展数据图 6f,附表 9)。利用染色体片段代换系 R3、R170(BC₅F₅)验证功能:两份株系将野生大豆 ZYD00006(Hap1)的YUC4a 片段导入栽培种绥农 14(Hap2),籽粒体积与百粒重均显著低于轮回亲本绥农 14(扩展数据图 6g、图 4g、4h)。综上,YUCCA4 基因在大豆、木豆、鹰嘴豆、豌豆驯化中发生趋同选择,正向驱动籽粒增重 。
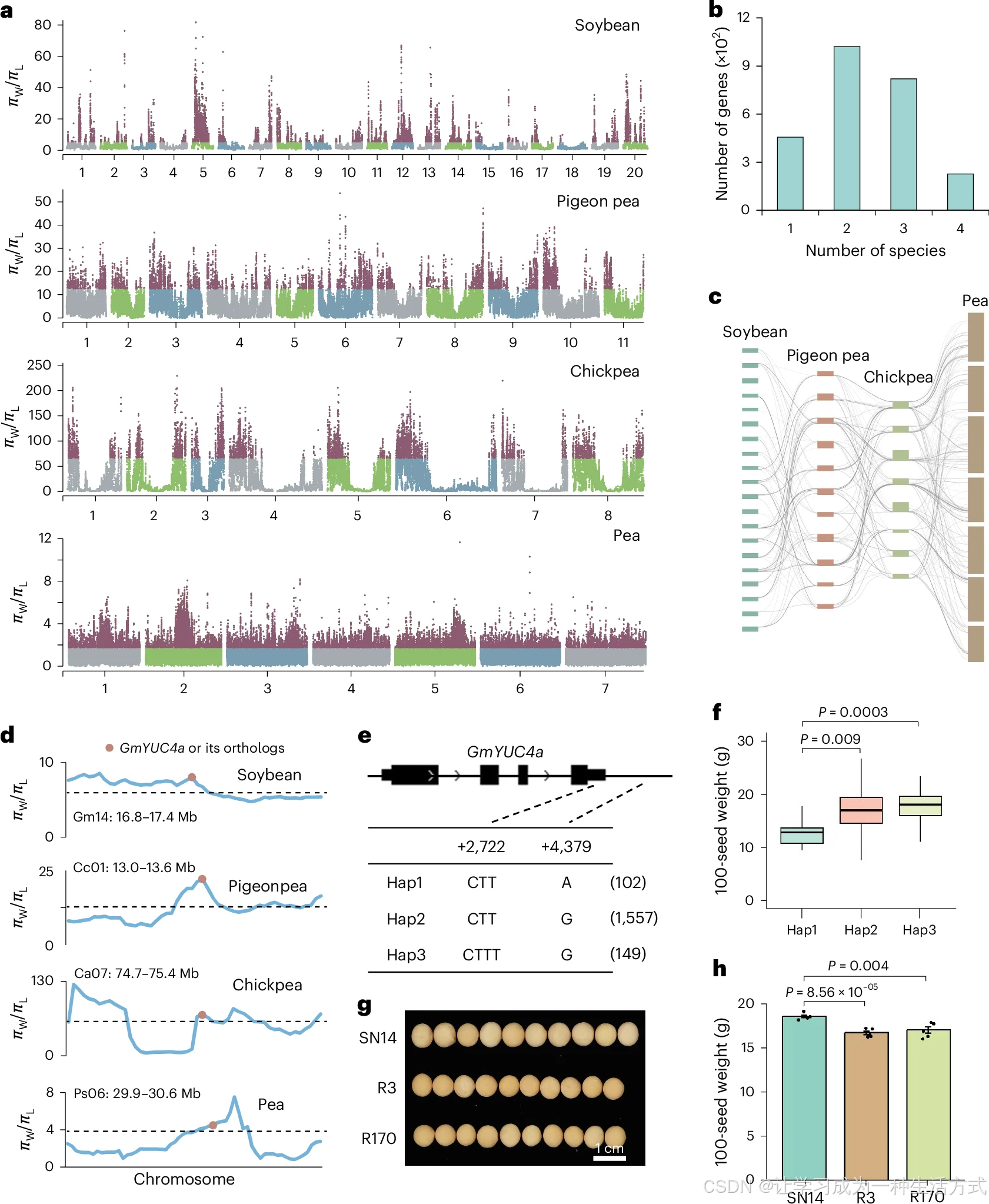
a. 大豆、木豆、鹰嘴豆、豌豆野生种与地方品种全基因组核苷酸多态性比值;\(\boldsymbol{\pi_W}\)代表野生群体 π 值,\(\boldsymbol{\pi_L}\)代表地方品种群体 π 值。b. 大豆受人工选择基因分类统计:1 为大豆特有驯化选择基因;2、3 分别代表大豆与另外 1 种、2 种豆类(木豆 / 鹰嘴豆 / 豌豆)共同趋同驯化基因;4 为四个物种同步趋同选择基因。c. 四类豆类趋同选择基因的基因组共线性排布。d. 大豆GmYUC4a 及其木豆CcYUC4 、鹰嘴豆CaYUC4 、豌豆PsYUC4 直系同源基因上下游各 600 kb 区间内,野生 / 地方品种 π 比值变化;采用100 kb 滑窗、10 kb 步长 计算。e. 大豆GmYUC4a 单倍型分型结果。f. Hap1(6 份材料)、Hap2(807 份)、Hap3(56 份)种质百粒重统计;双尾 t 检验统计显著性。箱线上下框线为 75%、25% 分位数,中线是中位数,须线延伸至 1.5 倍四分位距(IQR)。g. 栽培种绥农 14(SN14)与两份染色体片段代换系 R3、R170 籽粒表型实拍。h. 绥农 14、R3、R170 百粒重数据,每组 5 个生物学重复,结果以平均值 ± 标准差表示,显著性采用双尾 t 检验;SN14:绥农 14。
转座子局部扩增驱动基因组膨大
转座子(TE)扩张是塑造基因组演化的关键驱动力。暖季豆类整体转座子扩增幅度温和,插入富集在染色体中间区段(扩展数据图 8a、附表 3);而冷季豆类发生大规模转座子爆发式扩增(附表 3)。冷季豆类常染色质区转座子富集异常突出,占比 48%~90% (图 5a),异染色质区域转座子占比则稳定维持在约 90%(扩展数据图 8b)。无论常、异染色质区,长末端重复反转座子(LTR),尤其 Gypsy 超家族 的扩增幅度显著高于其他类型转座子(图 5a、扩展数据图 8b);其中蚕豆基因组转座子沿整条染色体均匀分布(图 5b)。
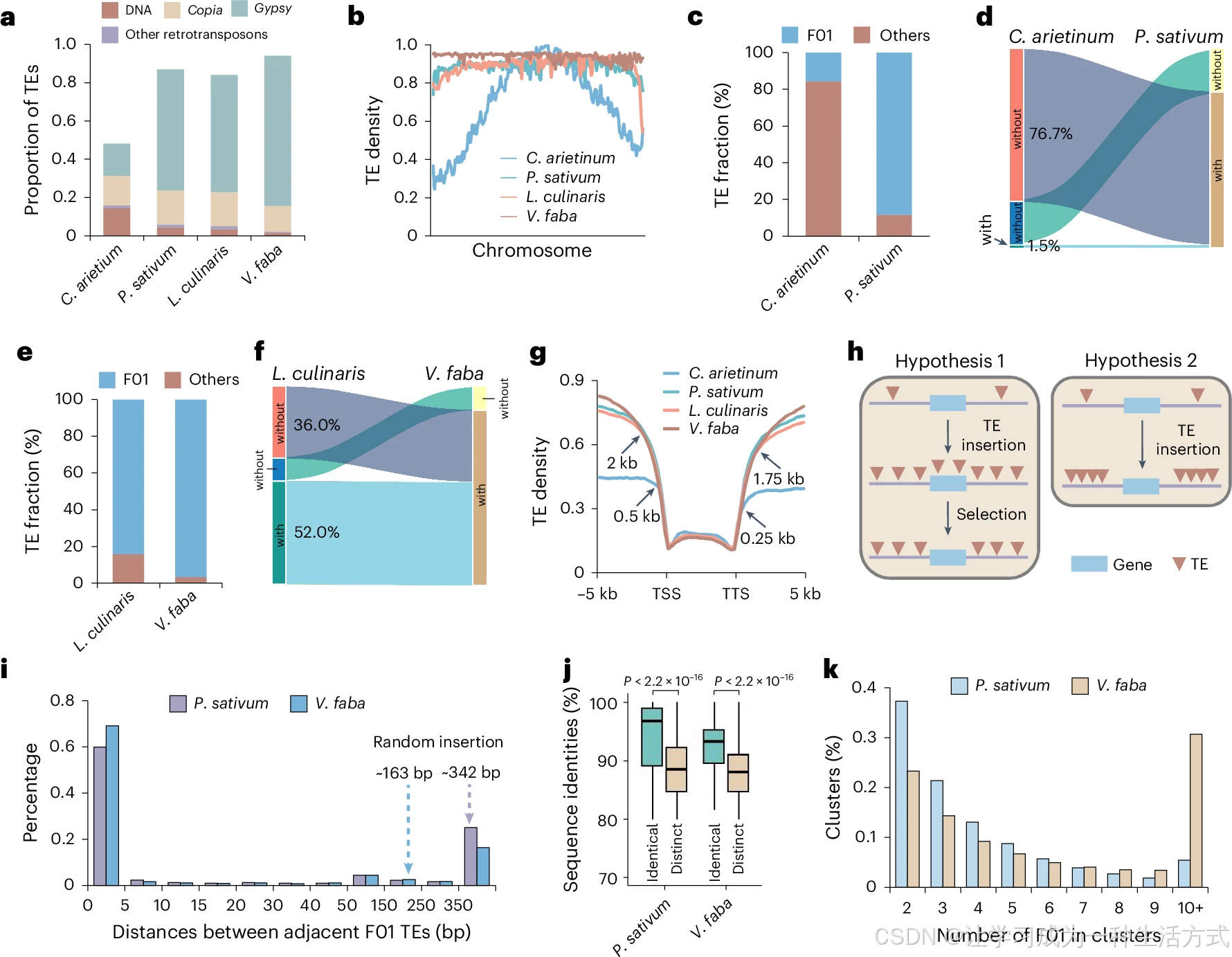
a. 常染色质区域中转座子(TE)的占比。b. 转座子在整条染色体上的分布;X 轴为染色体坐标,从染色体起始端到末端依次排布。c. 豌豆与鹰嘴豆 Gypsy 反转座子中 F01 亚型转座子的占比;分析区间选用两物种相邻成对基因间的共线性区段。d. 鹰嘴豆、豌豆基因组内,含 / 不含 F01 转座子的共线性区段占比。e. 小扁豆与蚕豆 Gypsy 元件中 F01 转座子占比;以两物种相邻成对基因间共线性区域作为分析背景。f. 小扁豆、蚕豆基因组中携带 / 不携带 F01 转座子的共线性区段占比。g. 四种冷季豆类保守基因上下游附近转座子的分布特征。h. 豆科演化中转座子扩张的两种假说:假说 1 代表转座子在基因组随机插入;假说 2 代表转座子偏好特定位点、成簇插入。i. 豌豆与蚕豆基因组中相邻 F01 转座子之间的间距分布。j. 同一簇内(豌豆\(n=48173\)、蚕豆\(n=328793\))与不同区域间(豌豆\(n=9676037\)、蚕豆\(n=57915589\))F01 转座子的序列一致性;采用双侧威尔科克森秩和检验计算P值。箱型图上下边框为 75%、25% 分位数,中间横线为中位数,须线延伸至 1.5 倍四分位距(IQR)。k. 不同拷贝数 F01 转座子簇的占比统计;间距≤5 bp 的相邻转座子合并划分为同一个插入簇。
为阐明基因组扩张过程中转座子的扩增模式,本研究筛选鹰嘴豆与其近缘物种豌豆间3830 个共线性区段 :鹰嘴豆区段平均长 9.6 kb,豌豆同源区段平均 62.6 kb;同理筛选小扁豆与近缘蚕豆共线性区段 3185 个,小扁豆平均 39.7 kb、蚕豆平均 155.8 kb。依托 80-80-80 分类标准,在共线性区间内注释得到 10 个主要转座子家族,其中F01 家族转座子 在冷季豆类中拷贝数最高(扩展数据图 8c~f、附表 10)。F01 与 Ty3-gypsy 型 Ogre 元件同源性最高,Ogre 也是豌豆、蚕豆基因组中丰度最高的转座子类型。F01 占豌豆全部 Gypsy 转座子的 88%,但在鹰嘴豆 Gypsy 家族中占比不足 16%(图 5c)。鹰嘴豆→豌豆的基因组区间拉长与 F01 插入扩张显著相关(扩展数据图 8g);鹰嘴豆中约 76.7% 不含 F01 的共线性区段,在豌豆同源区段均发生 F01 插入(图 5d)。小扁豆与蚕豆间存在相同规律:F01 在 Gypsy 中占比由小扁豆 85% 升至蚕豆 96%,伴随基因间区大幅扩张(扩展数据图 8h);小扁豆 36% 无 F01 的共线性区段在蚕豆中新增 F01 插入,超 50% 区段在两物种中均携带 F01(图 5f)。以上结果表明:鹰嘴豆演化至豌豆的基因组膨大以 F01 全新插入为主;小扁豆向蚕豆演化的基因组扩张则更多依靠原有 F01 在原位串联扩增 。
尽管冷季豆类基因组伴随大规模转座子扩增,但转座子极少插入基因本体及上下游 250 bp 侧翼区(图 5g),现有两种假说解释该特征:①转座子随机插入,插入基因区的拷贝受自然选择逐步淘汰;②转座子存在位点偏好、成簇聚集插入(图 5h)。通过统计相邻 F01 间距验证假说:豌豆约 60%、蚕豆约 70% 的相邻 F01 间距小于 5 bp,远低于随机插入模拟间距(豌豆均值 342 bp、蚕豆均值 163 bp,\(P<5×10^{-169}\),威尔科克森秩和检验),支持偏好性成簇插入假说 (图 5i)。同一基因间区内的 F01 序列一致性显著高于不同区间的 F01(图 5j);蚕豆的大片 F01 插入簇所占比例高于豌豆(图 5k)。为佐证转座子串联扩增规律,选取基因组大小与 TE 含量差异悬殊的二穗短柄草(271 Mb)、普通小麦(14.5 Gb)做参照:小麦 F01 丰度远高于短柄草,F01 插入中位间距 410 bp,远小于随机模拟值 4171 bp(\(P<5×10^{-117}\))。综上,基因贫瘠区中转座子的串联扩增是冷季豆类基因组膨大的重要驱动力,但不能完全排除随机插入 + 基因区转座子受选择删除的作用 。
相较于鹰嘴豆,豌豆、小扁豆、蚕豆染色体臂区段 DNA 甲基化整体显著升高,且甲基化沿染色体均匀分布,与蚕豆已有报道一致(扩展数据图 8k)。F01 转座子整体 CG 甲基化水平极高,仅不到 3.5‰的拷贝发生表达,和沉默型转座子修饰特征相符(扩展数据图 8l、m)。
调控元件的演化
转座子可通过序列变异演化为基因组调控元件。利用 ATAC-seq 染色质开放测序,在鹰嘴豆、豌豆、小扁豆、蚕豆分别鉴定开放染色质区域(OCR)22507、63137、56696、98458 个。按与转座子重叠关系分为三类:cTE-OCR(完全位于 TE 内部)、pTE-OCR(与 TE 部分重叠)、nTE-OCR(无 TE 重叠,附表 11)。cTE-OCR 占比从鹰嘴豆 0.8% 上升至蚕豆 10%(图 6a);相较于 pTE-OCR、nTE-OCR,cTE-OCR 更多富集在基因远端调控区(图 6b、扩展数据图 9a~c)。豌豆、小扁豆、蚕豆的 cTE-OCR 所在区段甲基化水平整体偏高,鹰嘴豆无此规律(图 6c、扩展数据图 9d~k)。玉米研究表明:近 20% 远端开放染色质来源于转座子,且 TE 内 OCR 区域甲基化显著低于周边侧翼序列。为验证豆类 TE 产生 OCR 是否伴随局部去甲基化,将含 OCR 的转座子拆分为 OCR 开放区与周边非 OCR 沉默区(图 6d、扩展数据图 9l、m)。结果与玉米一致:鹰嘴豆 TE 内部 OCR 区段甲基化显著低于相邻非 OCR 区;豌豆、小扁豆、蚕豆 TE 内 OCR 与侧翼非 OCR 甲基化无明显差异 ,说明后三者转座子形成开放染色质不依赖局部 DNA 去甲基化。
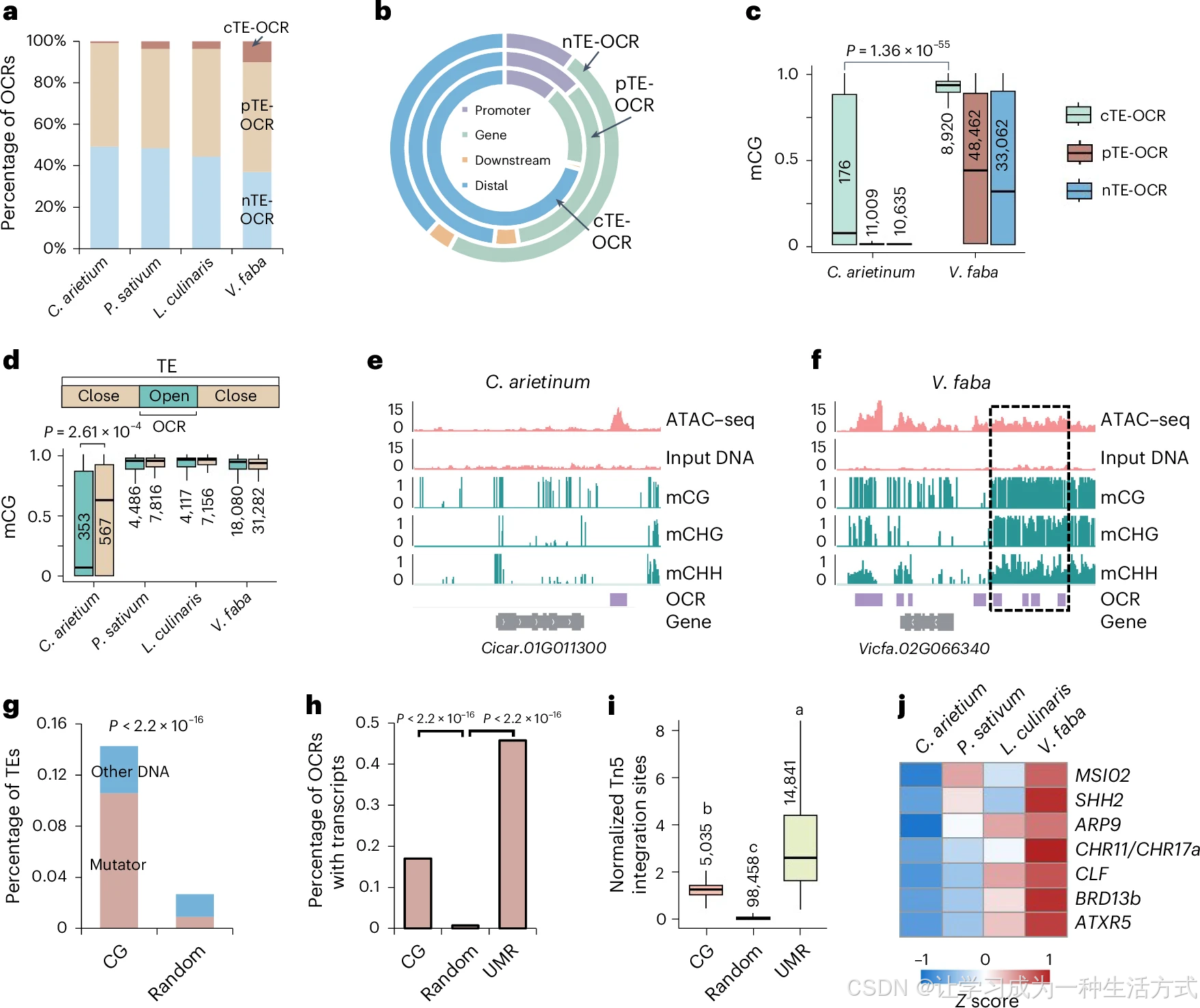
a. 完全转座子来源开放染色质(cTE-OCR)、部分转座子重叠开放染色质(pTE-OCR)、非转座子来源开放染色质(nTE-OCR)的占比。b. 蚕豆中三类 OCR 在基因本体、启动子区(转录起始位点上游 2 kb)、基因下游区(转录终止位点下游 500 bp)、远端基因间区的分布占比。c. 鹰嘴豆与蚕豆中 cTEOCR、pTEOCR、nTEOCR 的 DNA 甲基化水平。d. 转座子内部 OCR 开放区段与转座子其余闭合区段的 CG 甲基化水平。c、d 图注说明 :图中标注数字为样本量,显著性采用双侧威尔科克森秩和检验。e、f. IGV 基因组浏览器可视化截图,分别展示鹰嘴豆 (e)、蚕豆 (f) 中存在甲基化与无甲基化修饰的 OCR 区域。g. 蚕豆远端 CG 型 OCR 所重叠转座子中 DNA 转座子的占比;随机选取带 CG 甲基化的远端基因组区段作为对照组。h. 蚕豆中可检测到转录本的 CGOCR 与 UMROCR 占比。g、h 图注说明 :统计学显著性采用双侧费希尔精确检验。i. 蚕豆 CGOCR 与 UMROCR 的染色质开放程度,标注数字为样本量;单因素方差分析结合 Scheffe 多重比较检验差异显著性。j. 鹰嘴豆、豌豆、小扁豆、蚕豆中调控染色质可及性相关基因的表达量变化。箱线统一说明(c、d、i):箱体上下边界为 75%、25% 分位数,中间横线为中位数,须线延伸至 1.5 倍四分位距(IQR)。
已有研究表明,玉米、水稻等植物绝大多数开放染色质区域(OCR)位于非甲基化区(UMR)内。本研究结果与之吻合:鹰嘴豆超过 92% 的 OCR 落在非甲基化区段,定义为UMROCR (扩展数据图 10a);而豌豆、小扁豆、蚕豆中不足半数 OCR 与非甲基化区重叠(扩展数据图 10a)。除 UMROCR 外,在豌豆、小扁豆、蚕豆中分别鉴定出 5833、7713、11607 个高 CG 甲基化的开放染色质(CGOCR,附表 12)。例如,基因Cicar.01G011300 侧翼 OCR 在鹰嘴豆中甲基化水平很低(图 6e);但其蚕豆同源基因Vicfa.02G066340 周边大部分 OCR 呈现高甲基化修饰(图 6f)。约 28%~45% 的 CGOCR 定位于基因组远端区间,该区域显著富集 DNA 转座子,尤以 Mutator 超家族为主(图 6g、扩展数据图 10b~d)。相较于随机基因组区段,远端 CGOCR 拥有显著更高的转录活性与染色质开放度(图 6h、i,扩展数据图 10e~h)。本研究还筛选到 7 个染色质调控相关基因,其表达量从鹰嘴豆→豌豆 / 小扁豆→蚕豆呈现阶梯式上调(图 6j)。高甲基化 OCR 的物种分化,或与染色质调控因子的物种差异化表达密切相关。
讨论
作物驯化演化普遍伴随落粒性丧失、种子休眠减弱、籽粒变大与产量提升。目前已在多个物种中克隆一批跨物种平行驯化基因,如调控种子休眠的G 家族基因、调控穗粒数的KRN2 ;但全基因组水平系统筛选豆科跨物种趋同选择基因的研究仍较匮乏。本研究筛选得到 226 个在大豆、木豆、鹰嘴豆、豌豆中同步发生驯化型遗传多样性衰减的基因(图 4a~c)。从野生种到地方品种遗传多样性下降的基因,成因可分为人工驯化选择、环境适应性进化或遗传漂变。深入解析该类基因,既可为豆类种质改良提供理论支撑,也能为未来新型作物从头驯化、保障粮食安全提供参考。
转座子大规模扩增是豆科基因组膨胀的关键驱动力。LTR 反转座子是豌豆、蚕豆基因组最主要组成组分,其中 Ty3gypsy 型 Ogre 元件占比最高;但该类元件偏好富集在基因贫瘠区、回避基因富集区的扩增调控机制尚不清晰。本研究提出转座子串联扩增模型 :转座子通过识别基因组邻近保守基序实现定点插入,进而在局部聚集成簇(图 5h~k)。该成簇插入特征同样在小麦中被验证(扩展数据图 8j),果蝇中也有相关报道,说明该扩增机制在动植物中演化保守,但其分子机理仍需后续深入挖掘。除驱动基因组扩张外,转座子可逐步变异形成调控元件(图 6a),后续可通过调控元件敲除等功能试验,明确其在植物环境适应性中的生物学功能。传统观点认为植物调控区开放染色质普遍低甲基化;但本研究发现,基因组偏大的冷季豆类中相当一部分 OCR 呈现高甲基化修饰(图 6c~f、扩展数据图 9d~m)。玉米、小麦转座子占比同样超过 80%,但二者绝大多数 OCR 位于非甲基化区,由此说明:冷季豆类高甲基化 OCR 无法单纯用转座子扩张解释。
本研究仅选取全球主栽食用豆类开展解析,后续可扩充远缘属物种与同物种不同生态型种质,完善豆科演化图谱。将比较基因组与群体基因组、转录组联合分析,是挖掘农艺性状关键变异的重要方向。综上,本研究丰富了豆科演化理论,同时为豆类遗传育种提供高质量基因组资源。
试验方法
植物材料与测序
选取 9 种食用豆类开展基因组组装与注释:菜豆(苏菜豆 1917)、鹰嘴豆(XJ01)、豌豆(苏豌豆 08)、小扁豆(SX01)、蚕豆(P17186)、木豆(SM01)、豇豆(苏豇豆 01)、绿豆(苏绿豆 07)、藊豆(边红 01)。材料置于恒温 25 ℃、12 h 光照 / 12 h 黑暗培养箱育苗,取幼嫩叶片提取高分子量基因组 DNA。采用 PacBio 官方 SMRTbell Express Template Prep Kit 2.0 试剂盒构建插入片段约 15 kb 的 PacBio HiFi 文库,在 Sequel IIe 平台测序;鹰嘴豆额外利用 UltraLong DNA Sequencing Kit 构建平均插入~50 kb 的 ONT 超长片段文库,于 PromethION 平台测序。
9 个物种均参照已有方法构建 HiC 染色质构象捕获文库,Illumina NovaSeq 6000 平台测序,产出 150 bp 双端 reads。
转录组测序:分别采集萌发 20 d 幼苗幼叶与茎、播种 15 d 根系、授粉 20 d 豆荚组织,Invitrogen TRIzol 试剂提取总 RNA;Vazyme VAHTS 通用 RNA 建库试剂盒构建转录组文库,NovaSeq 6000 测序,产出 150 bp 双端数据,用于后续基因结构注释。
基因组组装
基因组组装参照已有文献流程开展。简要流程:在 SMRT Link v10.1 软件中调用 ccs(v6.0.0),设置参数--min-passes 3生成 HiFi 高精度 reads;利用 hifiasm(v0.16.1)默认参数完成基因组初步组装。筛选长度>15 kb、Phred 质控值>7 的 ONT 超长读段,借助 minimap2(v2.24,参数-x ava-ont)+ miniasm(v0.3,参数-Rc2)组装 ONT contig。针对鹰嘴豆:依托 RagTag(v2.1.0)的 scaffold 模块,以 ONT 组装 contig 为骨架锚定 PacBio HiFi contig;再用 purge_dups(v1.2.5)剔除 contig 中残留杂合冗余拷贝。整合 HiC 数据挂载至染色体水平:HiC 原始数据经 TrimGalore(v0.6.4)默认参数去除接头与低质量 reads;合格 reads 用 Juicer(v1.6)流程比对基因组并去冗余,仅保留比对质量 MAPQ≥20 的互作片段;3DDNA(v190716)配置--mapq 20校正组装错误、完成 contig 排序与定向挂载;最终在 Juicebox 软件中依托 HiC 互作图谱人工微调,无法锚定的零散片段单独保留为独立 scaffold。
基因组注释
转座子注释
转座子从头注释分两步:分别用 LTRharvest(v1.6.2)、LTR_FINDER_parallel(v1.1)预测全长 LTR 元件,结果经由 LTR_retriever(v2.9.0)去冗余,构建非冗余 LTR 参考库;同时使用 RepeatModeler(v2.0.1)从头构建全基因组重复序列库,未知分类转座子通过 DeepTE 完成分类。最终调用 RepeatMasker(v4.1.2),将基因组序列比对 LTR 库、自建重复库与 Repbase (2018) 数据库,完成全基因组 TE 屏蔽注释。
基因结构注释
整合转录组证据、同源蛋白比对、从头预测 三套结果进行基因模型构建:
- 转录组证据 :多组织 RNAseq 数据经 HISAT2(v2.1.0)比对基因组,StringTie(v2.1.4)基于比对结果拼接转录本;同时 Trinity(v2.14.0)无参组装转录本,两套转录产物经由 PASA(v2.5.2)比对基因组优化基因结构,TransDecoder 预测候选编码区。
- 同源预测 :采用 GeMoMa(v1.8),依托 14 个已发表物种蛋白序列(栽培大豆、狭叶大豆、鹰爪大豆、菜豆、豇豆、白羽扇豆、绿豆、木豆、鹰嘴豆、豌豆、蒺藜苜蓿、番茄、水稻、拟南芥)进行同源基因定位。
- 从头预测 :使用 Augustus(v3.3.3)进行基因从头预测。三套基因模型通过 EVidenceModeler(v1.1.1)整合得到非冗余基因集,再经过两轮 PASA 迭代修正;利用 BUSCO(v5.4.2)基于通用单拷贝同源基因评估组装完整度;Interproscan (5.6092.0) 配置-appl PRINTS,Pfam,SMART,PANTHER,ProSiteProfiles,SUPERFAMILY -goterms注释蛋白保守结构域与 GO 功能。
基因家族演化分析
以本研究 9 个新组装基因组 + 大豆、野生大豆、白羽扇豆共 12 个豆科基因组构建泛基因组,OrthoFinder(v2.5.4)默认参数聚类得到层级同源基因簇;按照泛基因组标准将基因家族划分为核心、软核心、壳、物种特有四类。基于 WGDI(v0.6.2)内置 YN00 算法计算核心 / 软核心 / 壳基因的非同义替换率 Ka、同义替换率 Ks;OrthoFinder 配置-M msa -A mafft对单拷贝基因做多序列比对,iqtree(v2.2.0.3,-B 1000 --bnni)依托比对文件构建物种进化树,拟南芥作为外类群定根。利用 PAML(v4.10.6)中 MCMCTree,结合 TIMETREE 化石校正数据估算物种分化时间并构建超度量进化树;CAFE5 (v5.0.0,\(P<0.05\)) 分析各分支基因家族扩张 / 收缩;进化树用 iTOL(Interactive Tree of Life)可视化。
结瘤相关基因分析
选用大豆作为模式物种解析结瘤相关基因演化规律。收集已发表大豆根瘤、根系及不同发育时期其他组织转录组数据,共计 7 组根瘤样本、33 组其他组织样本(附表 13)。原始测序数据经 TrimGalore (v0.6.8) 去除接头与低质量读段;干净 reads 利用 HISAT2 (v2.1.0,参数--dta --no-discordant --no-mixed) 比对至大豆 Wm82-NJAU 参考基因组;仅保留 MAPQ≥20 的唯一比对 reads,通过 Stringtie (v2.1.4,参数-e -B) 定量基因表达。根瘤偏好表达基因筛选标准 :①至少在 2 个根瘤样本中 FPKM≥1;②至少 2 个根瘤样本的表达量≥任意非根瘤组织表达量的 2 倍。
依据物种进化树溯源基因起源节点,将大豆全部基因划分为 0~7 共 8 个演化等级:0 级为大豆物种特有基因(最晚起源);7 级为大豆与葡萄共同祖先起源、跨物种保守度最高的基因。
固氮分支 (NFC) 新生起源基因筛选
筛选在 12 个豆科全物种存在、11 个外类群(玉米、水稻、小麦、番茄、葡萄、毛果杨、拟南芥、陆地棉、核桃、黄瓜、桃)全部缺失的基因 / 基因家族;再通过 BLAST 剔除与任意外类群基因同源\(E<1×10^{-10}\)的序列,剩余候选基因为新生基因。
- 在木麻黄(壳斗目)、大卫羊蹄甲(葫芦目)、沙棘(蔷薇目)中存在同源:归为固氮分支 NFC 演化阶段诞生的新生基因 ;
- 在紫荆(云实亚科)、含羞草(云实亚科)中存在同源:归为豆目分化时诞生的新生基因 ;
- 其余:在蝶形花亚科豆科祖先阶段新生。
NLR 抗病基因鉴定与分型
沿用已报道流程筛选 NLR 基因:筛选氨基酸长度≥150 的蛋白序列,hmmsearch (v3.3.1,-E 1e-10) 搜索 NBARC 保守结构域(PF00931.25);同时 Interproscan (5.6092.0,-appl Pfam, Coils) 注释 TIR、RPW8 类卷曲螺旋 (CCR)、普通 CC、NBARC、LRR 结构域。依据结构域组成将 NLR 分为四类:
- TNL:含 TIR 结构域;
- CNL:同时含 CC+NBARC;
- RNL:含 RPW8 结构域;
- NL:仅含 NB 结构域。
WGD/SSD/ 单拷贝基因分类
各物种蛋白序列自比对(BLAST,\(E<1×10^{-10}\)),保留相似度与比对覆盖度均≥70% 的比对结果;调用 MCScanX 的duplicate_gene_classifier模块,把基因分为:全基因组复制来源基因 (WGD)、小规模复制基因 SSD(串联复制、邻近复制、散在复制)、单拷贝基因 (singleton)。
驯化选择位点挖掘与趋同选择基因鉴定
收集大豆、木豆、鹰嘴豆野生祖先种与地方品种重测序数据,豌豆野生种质与栽培种重测序数据(附表 14)。质控后 reads 经 BWA (v0.7.17) 比对参考基因组,Picard (v2.27.4) 去 PCR 重复,GATK (v4.1.3.0) HaplotypeCaller 挖掘 SNP;PLINK (v1.9) 筛选:最小等位基因数≥5、缺失率≤0.2 的高质量 SNP。全基因组采用100 kb 滑窗、步长 10 kb 分箱,VCFtools (v0.1.16) 计算每窗群体核苷酸多态性 π;利用公式\(\boldsymbol{ROD=1-\pi_{地方种}/\pi_{野生种}}\)计算多样性衰减系数。ROD 全群体分布前 90% 区间 判定为受选择清除区段;在四大豆类受选择区间内均保守存在的同源基因为跨物种趋同驯化基因 (附表 7、附表 8)。YUC1/YUC4 基因上下游区间同样采用 100 kb 滑窗、10 kb 步长,VCFtools 计算群体分化系数\(F_{ST}\)。
Gypsy 类转座子分型
依托808080 规则 划分 Gypsy 转座子家族:筛选长度≥80 bp 的 Gypsy 序列做 BLAST 自比对(\(E<1×10^{-10}\));序列相似度≥80%、比对覆盖度≥80% 的序列归为同一组;单组成员≥100 条转座子方可认定为有效家族。
MethylC-seq 建库与数据分析
选取鹰嘴豆、豌豆、小扁豆、蚕豆、菜豆、豇豆、绿豆、藊豆、木豆、野生大豆、栽培大豆共计 11 个豆科物种,每个物种设置2 个生物学重复 ,构建全基因组重亚硫酸盐甲基化测序(MethylCseq)文库。采用 CTAB 法提取幼叶基因组 DNA,Covaris M220 超声破碎为 300~500 bp 片段;使用 NEB NEBNext Ultra II 试剂盒完成末端修复、加 A 尾、甲基化接头连接;Zymo EZ DNA MethylationGold 试剂盒进行重亚硫酸盐转化;PCR 扩增 13~15 个循环后,Vazyme 磁珠纯化文库,Illumina NovaSeq 平台上机测序。
原始数据经去接头与低质量过滤后,Bismark (v0.23.1) 默认参数比对对应参考基因组;仅保留唯一比对读段,比对至同一位置的多条读段合并为一致性序列以降低 PCR 扩增偏好;分段统计基因组各区段加权甲基化水平;利用 λDNA 计算各文库重亚硫酸盐转化率。
ATAC-seq 建库与数据分析
上述 11 个物种幼叶取材,各设 2 个生物学重复,参照已发表方案构建染色质开放测序 ATACseq 文库。取约 0.2 g 新鲜叶片置于含 3 mL 预冷裂解液的培养皿中剪碎(裂解液组分:25 mM TrisHCl、0.44 M 蔗糖、10 mM MgCl₂、0.1% Triton X100、2 mM 精胺、1 mM PMSF、1× 罗氏蛋白酶抑制剂、10 mM β巯基乙醇);40 μm 细胞筛过滤,细胞核先后用细胞核提取缓冲液 1 洗涤 2 次、提取缓冲液 2 洗涤 1 次;纯化后的细胞核重悬于提取缓冲液 1 并镜检完整性;取约 50000 个细胞核,加入 Vazyme Tn5 转座酶 37 ℃孵育 30 min,后续建库、NovaSeq 平台 150 bp 双端测序;每个物种同步以基因组裸 DNA 构建阴性对照文库。
测序数据经 TrimGalore (v0.6.8,参数--stringency 3 --trim-n --max_n 7) 去除接头与劣质读段;Bowtie2 (v2.2.9,-X 1000) 比对基因组;Picard (v2.27.4) 去除 PCR 重复,保留 MAPQ≥20 的正确配对比对序列。为消除测序深度偏差,使用 Picard 将 ATACseq 与对照裸 DNA 文库测序量下采样至同一水平;MACS2 (v2.2.7.1,--nomodel --shift -100 --extsize 200 -f BAM -q 0.01 --keep-dup all) 鉴定开放染色质区域(OCR);依据 OCR 与邻近转座子的重叠关系分为三类 OCR。
非甲基化区(UMR)与高 CG 甲基化区域鉴定
全基因组无重叠划分为 100 bp 连续窗口,仅保留每种甲基化模式下胞嘧啶测序深度≥2 且至少含 2 个胞嘧啶的区段;CG/CHG/CHH 三类甲基化水平均<10% 的区段定义为非甲基化区 UMR ;CG 甲基化>90% 的区段为高 CG 甲基化区;若单个 OCR 超过 60% 区间被高 CG 甲基化区覆盖,则该 OCR 归类为 CGOCR。
统计学分析与试验可重复性说明
所有统计检验类型、样本量n详见图 1、图 4~图 6 图注;本研究泛基因组分析未通过统计学方法预先设定样本量,分析过程无数据剔除;试验未采用随机分组设计,试验人员在试验分组与结果统计环节未实施盲法。