一、写在前面
经常有同学咨询入手足够完成硕博生涯的生信环境能否完成一百万个单细胞的数据分析,其实这个问题比较难回答,大家的数据质量、分析环境、需要做的分析、算法都不尽相同。很难准确的评估。不过大概的量级还是可以测评的,我们往期也测试过1~30w细胞级别单细胞数据的硬件占用情况:
30w单细胞数据会吃掉多少内存?
5w单细胞在Monocle2拟时序中占据多少内存?
CellChat细胞通讯运行内存及时间压力测试
这里,我们干脆就以GSE184652这个数据为例,给大家演示真实的百万级数据集在基础分析、多样本整合、细胞通讯、拟时序分析分析过程中分别需要多少内存 。不过从省流的角度而言,推荐直接抛弃R语言,考虑scRNA-Seq学习手册Python版。
本教程基于Linux环境演示,计算资源不足的同学可参考:
足够支持你完成硕博生涯的生信环境
忘记宣传了,独享用户连技术支持都是独享的
如果你对下面的教程比较迷茫,那么你可以先行学习编程教程:
二、大算特算
# 先记录一下运行前的时间:
Sys.time()
## [1] "2025-08-31 07:16:49 UTC"1、测试数据
#### 加载需要的R包 ####
suppressMessages({
library(Seurat)
library(CellChat)
library(peakRAM)
library(patchwork)
library(RColorBrewer)
library(colorspace)
library(biovizBase)
})
#设置并行计算
makecore <-function(workcore,memory){
if(!require(Seurat))install.packages('Seurat')
if(!require(future))install.packages('future')
plan("multisession", workers = workcore)
options(future.globals.maxSize= memory*1024*1024**2)
}
makecore(2,100)#这里以四线程,50GB为例
## Loading required package: future
##
## Attaching package: 'future'
## The following objects are masked from 'package:igraph':
##
## %->%, %<-%
# 读取数据(光读取就要很久):
scRNA_count <-Read10X('./data/GSE184652_count/',gene.column =1)
# 细胞数量,这个数据被作者过滤过,不过也妥妥的是百万级的数据集了:
ncol(scRNA_count)
## [1] 9466602、基础分析
开始做经典分析,看看做到降维分群一共需要花费多少的时间以及计算资源,解读和教程可见单样本分析。peakRAM 是 R 语言 peakRAM 包中的一个核心函数,专门用于精确监控和记录 R 代码执行过程中的内存使用情况,特别是峰值内存占用。
peakRAM({
scRNA <-CreateSeuratObject(counts = scRNA_count, project ="scRNA_million", min.cells =3, min.features =200)
meta <-read.csv(gzfile('data/GSE184652_metadata.csv.gz'),row.names =1)
scRNA <-AddMetaData(scRNA,metadata = meta)
scRNA[["percent.mt"]] <-PercentageFeatureSet(scRNA, pattern ="^MT-")
# scRNA <- subset(scRNA, subset = nFeature_RNA > 200 & nFeature_RNA < 2500 & percent.mt < 5) #已经不够一百万了,就先不过滤了
scRNA <-NormalizeData(scRNA, normalization.method ="LogNormalize", scale.factor =10000)
scRNA <-FindVariableFeatures(scRNA, selection.method ="vst", nfeatures =2000)
scRNA <-ScaleData(scRNA, features =VariableFeatures(scRNA))
scRNA <-RunPCA(scRNA, features =VariableFeatures(object = scRNA))
scRNA <-FindNeighbors(scRNA, dims =1:20)
scRNA <-FindClusters(scRNA, resolution =0.1)
scRNA <-RunUMAP(scRNA, dims =1:20)
scRNA <-RunTSNE(scRNA, dims =1:20)
scRNA.markers <-FindAllMarkers(scRNA, only.pos =TRUE, min.pct =0.25, logfc.threshold =0.25)
})
## Normalizing layer: counts
## Finding variable features for layer counts
## Centering and scaling data matrix
## PC_ 1
## Positive: Slc34a1, Slc4a4, Fut9, Ndrg1, Dab2, Nhs, Gk, Lrp2, Pter, Errfi1
## Abcc2, Pck1, Nox4, Acsm2, Slc13a1, Gldc, Ppara, Osbpl8, Ghr, Acox1
## Gramd1b, Keg1, Tnfaip8, Hsd3b2, Hnf4aos, Tnfrsf21, Glud1, Slc27a2, Chpt1, Fbp1
## Negative: Mecom, Dach1, Stk39, Stox2, Pde7b, Slit2, Maml2, Cacnb4, Pde10a, Prdm16
## Slc16a7, Dnm3, Tacc1, Abca13, Phactr1, Nudt4, Iqgap1, Erbb4, Efna5, Kctd1
## Utrn, Ccser1, Nr3c2, Sorbs2, Sim1, Kng2, Slc8a1, Hk1, Wnk1, Efhd1
## PC_ 2
## Positive: Meis2, Ldb2, Ebf1, Dlc1, Prkg1, Plpp1, Tshz2, Eng, Cald1, Flt1
## Emcn, Cped1, Adgrl4, Heg1, Ptprb, Exoc3l2, Fgd5, Gm26883, Cyyr1, Rapgef5
## Arhgap31, Nrp2, Tek, Plpp3, Prex2, Hip1, Fbxl7, St6galnac3, B3galt1, Shank3
## Negative: Abca13, Cacnb4, Slc16a7, Kng2, Slit2, Chka, Mecom, Nudt4, Paqr5, Prdm16
## Kctd1, Galnt18, Oxct1, Slc8a1, Wnk1, Erbb4, Stk39, Tsc22d1, Defb1, Dach1
## Efhd1, Nckap5, Sim1, Sgms2, Lnx1, Me3, Phactr1, Tmem72, Kcnj1, Klhl3
## PC_ 3
## Positive: Flt1, Adgrl4, Exoc3l2, Ptprb, Cyyr1, Tek, Emcn, Shank3, Plpp1, Pecam1
## Rgcc, Rapgef4, Prkch, Fam117b, Samd12, Plvap, Dysf, Kdr, Ushbp1, Ccdc85a
## St6galnac3, Entpd1, Arhgap31, Lrrc8c, Plpp3, Gimap6, Inpp4b, Tll1, Cd300lg, Rasgrp3
## Negative: Lama2, Gm2163, 4930578G10Rik, Cfh, Hsd11b1, Csmd1, B3galt1, Rerg, Pde3a, Olfr1033
## Gucy1a2, Abca8a, Svep1, Kcnt2, Fhl2, Lhfp, Fbln5, Robo2, Ak5, Gpc6
## Clca3a1, Gm12394, Slco2b1, Pdgfra, Pcdh9, Adam12, Ctgf, Daam2, Mrc2, C7
## PC_ 4
## Positive: Slc5a12, Slc5a2, Slc7a7, Magi2, Nox4, Slc7a8, Gatm, Epb41l3, Gm37245, Slc16a10
## Adra1a, 4921539H07Rik, Car12, Slc16a14, Acss1, Tnfaip8, Slc34a1, Dab2, Slc4a4, Cyp24a1
## Agmo, Col27a1, Pah, Col8a1, Chd9, Angpt1, Slc13a1, Alpl, Unc5c, Timp3
## Negative: Napsa, Slc7a12, Aadat, 4930533I22Rik, Vmn1r18, Slc6a18, Rnf24, Gm15396, Mep1b, Acox2
## Mat2a, Slc22a19, Atp11a, Mep1a, Mpv17l, Gm42397, Mro, H3f3b, Wdr17, Serpina1f
## Gclm, Kcnc3, Slc5a4a, Gpm6a, Nudt19, Gm4450, Hao2, Gm6614, Adgrb3, Cbr1
## PC_ 5
## Positive: Frmpd4, Bmpr1b, Kif26b, Aqp2, Fam129a, Pde1c, Grem2, Mgat4c, Fxyd4, Naaladl2
## Scnn1b, Alcam, Cacnb2, Atp6v1g3, Tmem45b, Spink8, Scnn1g, Col26a1, Hsd11b2, Wscd2
## Atp6v0d2, Mpped2, Fanca, Rhcg, Lsamp, Slc4a9, Slc26a4, Iqgap2, Fgf12, Sdk1
## Negative: Egf, Esrrb, Slc12a1, Ppp1r1a, Abca13, Erbb4, Umod, Kng2, Fxyd2, Stk32b
## Ptger3, Prkd1, Ank2, Tmem72, Enox1, Gm14002, Slc2a13, Tenm2, Slc12a3, Cacnb4
## Lrrc66, Pcsk6, Sgcz, Bckdhb, Plekhg1, Lpl, Ckb, Pla1a, Tmem207, Gm42697
## Computing nearest neighbor graph
## Computing SNN
## Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
##
## Number of nodes: 946660
## Number of edges: 22156310
##
## Running Louvain algorithm...
## Maximum modularity in 10 random starts: 0.9752
## Number of communities: 26
## Elapsed time: 805 seconds
## 12 singletons identified. 14 final clusters.
## Warning: UNRELIABLE VALUE: One of the 'future.apply' iterations
## ('future_lapply-1') unexpectedly generated random numbers without declaring so.
## There is a risk that those random numbers are not statistically sound and the
## overall results might be invalid. To fix this, specify 'future.seed=TRUE'. This
## ensures that proper, parallel-safe random numbers are produced via the
## L'Ecuyer-CMRG method. To disable this check, use 'future.seed = NULL', or set
## option 'future.rng.onMisuse' to "ignore".
## Warning: The default method for RunUMAP has changed from calling Python UMAP via reticulate to the R-native UWOT using the cosine metric
## To use Python UMAP via reticulate, set umap.method to 'umap-learn' and metric to 'correlation'
## This message will be shown once per session
## 08:12:42 UMAP embedding parameters a = 0.9922 b = 1.112
## 08:12:42 Read 946660 rows and found 20 numeric columns
## 08:12:42 Using Annoy for neighbor search, n_neighbors = 30
## 08:12:42 Building Annoy index with metric = cosine, n_trees = 50
## 0% 10 20 30 40 50 60 70 80 90 100%
## [----|----|----|----|----|----|----|----|----|----|
## **************************************************|
## 08:14:21 Writing NN index file to temp file /tmp/Rtmpac0Got/file28f4284a253475
## 08:14:21 Searching Annoy index using 2 threads, search_k = 3000
## 08:18:40 Annoy recall = 100%
## 08:18:41 Commencing smooth kNN distance calibration using 2 threads with target n_neighbors = 30
## 08:19:06 Initializing from normalized Laplacian + noise (using RSpectra)
## 08:32:08 Commencing optimization for 200 epochs, with 43924856 positive edges
## 08:41:21 Optimization finished
## Calculating cluster 0
## Calculating cluster 1
## Calculating cluster 2
## Calculating cluster 3
## Calculating cluster 4
## Calculating cluster 5
## Calculating cluster 6
## Calculating cluster 7
## Calculating cluster 8
## Calculating cluster 9
## Calculating cluster 10
## Calculating cluster 11
## Calculating cluster 12
## Calculating cluster 13
## Function_Call
## 1 {scRNA<-CreateSeuratObject(counts=scRNA_count,project="scRNA_million",min.cells=3,min.features=200)meta<-read.csv(gzfile("data/GSE184652_metadata.csv.gz"),row.names=1)scRNA<-AddMetaData(scRNA,metadata=meta)scRNA[["percent.mt"]]<-PercentageFeatureSet(scRNA,pattern="^MT-")scRNA<-NormalizeData(scRNA,normalization.method="LogNormalize",scale.factor=10000)scRNA<-FindVariableFeatures(scRNA,selection.method="vst",nfeatures=2000)scRNA<-ScaleData(scRNA,features=VariableFeatures(scRNA))scRNA<-RunPCA(scRNA,features=VariableFeatures(object=scRNA))scRNA<-FindNeighbors(scRNA,dims=1:20)scRNA<-FindClusters(scRNA,resolution=0.1)scRNA<-RunUMAP(scRNA,dims=1:20)scRNA<-RunTSNE(scRNA,dims=1:20)scRNA.markers<-FindAllMarkers(scRNA,only.pos=TRUE,min.pct=0.25,logfc.threshold=0.25)}
## Elapsed_Time_sec Total_RAM_Used_MiB Peak_RAM_Used_MiB
## 1 14907.86 41092.2 123286
gc()
## used (Mb) gc trigger (Mb) max used (Mb)
## Ncells 14338991 765.8 23938628 1278.5 23938628 1278.5
## Vcells 7072585313 53959.6 17943776855 136900.2 17845893505 136153.4
# 保存数据:
saveRDS(scRNA,'data/scRNA.rds')
# 清楚环境变量和内存:
rm(list =ls());gc()
## used (Mb) gc trigger (Mb) max used (Mb)
## Ncells 14321067 764.9 23938628 1278.5 23938628 1278.5
## Vcells 5400291918 41201.0 17943776855 136900.2 17845893505 136153.43、细胞通讯
这里我们以CellChat为例,更多细胞通讯计算方法可参考一文学会四种scRNA-seq细胞通讯分析。需要注意的是,细胞通讯通常是先分别对各个组别拆分开进行细胞通讯计算,再在多组别细胞通讯中进行合并可视化。这意味着我们此次的演示数据单组别即达到百万数据集,那实战中大家可以完成数百万个单细胞数据集的细胞通讯计算。
try({
peakRAM({
# 加载所需R包
suppressMessages(if(!require(CellChat))devtools::install_github("sqjin/CellChat"))
suppressMessages(if(!require(ggplot2))install.packages('ggplot2'))
suppressMessages(if(!require(patchwork))install.packages('patchwork') )
suppressMessages(if(!require(ggalluvial))install.packages('ggalluvial'))
suppressMessages(if(!require(igraph))install.packages('igraph'))
suppressMessages(if(!require(dplyr))install.packages('dplyr'))
suppressMessages(options(stringsAsFactors =FALSE))
scRNA <-readRDS('data/scRNA.rds')
scRNA <-subset(scRNA, subset = celltype %in%names(which(table(scRNA$celltype) >0)))
scRNA <- scRNA[,!is.na(scRNA$celltype)]
scRNA <-JoinLayers(scRNA)
data.input <-GetAssayData(scRNA,'RNA','data')
meta <- scRNA@meta.data
cellchat <-createCellChat(object = data.input, meta = meta, group.by ="celltype")
cellchat <-addMeta(cellchat, meta = meta)
cellchat <-setIdent(cellchat, ident.use ="celltype")
groupSize <-as.numeric(table(cellchat@idents))
CellChatDB <- CellChatDB.mouse
CellChatDB.use <-subsetDB(CellChatDB, search ="Secreted Signaling")
cellchat@DB <- CellChatDB.use
cellchat <-subsetData(cellchat,features =NULL)
cellchat <-identifyOverExpressedGenes(cellchat)
cellchat <-identifyOverExpressedInteractions(cellchat)
cellchat <-computeCommunProb(cellchat, raw.use = T)
cellchat <-filterCommunication(cellchat, min.cells =10)
cellchat <-computeCommunProbPathway(cellchat)
cellchat <-aggregateNet(cellchat)
})
})
## Warning: Removing 12481 cells missing data for vars requested
## [1] "Create a CellChat object from a data matrix"
## Warning in createCellChat(object = data.input, meta = meta, group.by = "celltype"): The 'meta' data does not have a column named `samples`. We now add this column and all cells are assumed to belong to `sample1`!
## Set cell identities for the new CellChat object
## The cell groups used for CellChat analysis are CNT, DCT, DTL, EC, Fib, ICA, ICB, Immune, Inj.PT, JGA, MD, PC, PCT, PEC, Podo, PST, tAL, TAL
## Warning in asMethod(object): sparse->dense coercion: allocating vector of size
## 4.7 GiB
## The number of highly variable ligand-receptor pairs used for signaling inference is 950
## triMean is used for calculating the average gene expression per cell group.
## Warning in asMethod(object): sparse->dense coercion: allocating vector of size
## 4.7 GiB
## [1] ">>> Run CellChat on sc/snRNA-seq data <<< [2025-08-31 12:08:22.374398]"
## [1] ">>> CellChat inference is done. Parameter values are stored in `object@options$parameter` <<< [2025-08-31 12:59:35.413955]"
## Function_Call
## 1 {suppressMessages(if(!require(CellChat))devtools::install_github("sqjin/CellChat"))suppressMessages(if(!require(ggplot2))install.packages("ggplot2"))suppressMessages(if(!require(patchwork))install.packages("patchwork"))suppressMessages(if(!require(ggalluvial))install.packages("ggalluvial"))suppressMessages(if(!require(igraph))install.packages("igraph"))suppressMessages(if(!require(dplyr))install.packages("dplyr"))suppressMessages(options(stringsAsFactors=FALSE))scRNA<-readRDS("data/scRNA.rds")scRNA<-subset(scRNA,subset=celltype%in%names(which(table(scRNA$celltype)>0)))scRNA<-scRNA[,!is.na(scRNA$celltype)]scRNA<-JoinLayers(scRNA)data.input<-GetAssayData(scRNA,"RNA","data")meta<-scRNA@meta.datacellchat<-createCellChat(object=data.input,meta=meta,group.by="celltype")cellchat<-addMeta(cellchat,meta=meta)cellchat<-setIdent(cellchat,ident.use="celltype")groupSize<-as.numeric(table(cellchat@idents))CellChatDB<-CellChatDB.mouseCellChatDB.use<-subsetDB(CellChatDB,search="SecretedSignaling")cellchat@DB<-CellChatDB.usecellchat<-subsetData(cellchat,features=NULL)cellchat<-identifyOverExpressedGenes(cellchat)cellchat<-identifyOverExpressedInteractions(cellchat)cellchat<-computeCommunProb(cellchat,raw.use=T)cellchat<-filterCommunication(cellchat,min.cells=10)cellchat<-computeCommunProbPathway(cellchat)cellchat<-aggregateNet(cellchat)}
## Elapsed_Time_sec Total_RAM_Used_MiB Peak_RAM_Used_MiB
## 1 3736.449 53203.8 125972.4
# 清除环境变量和内存:
rm(list =ls());gc()
## used (Mb) gc trigger (Mb) max used (Mb)
## Ncells 13575303 725.0 23938628 1278.5 23938628 1278.5
## Vcells 26939077 205.6 13229728600 100934.9 16536802186 126165.84、拟时序分析
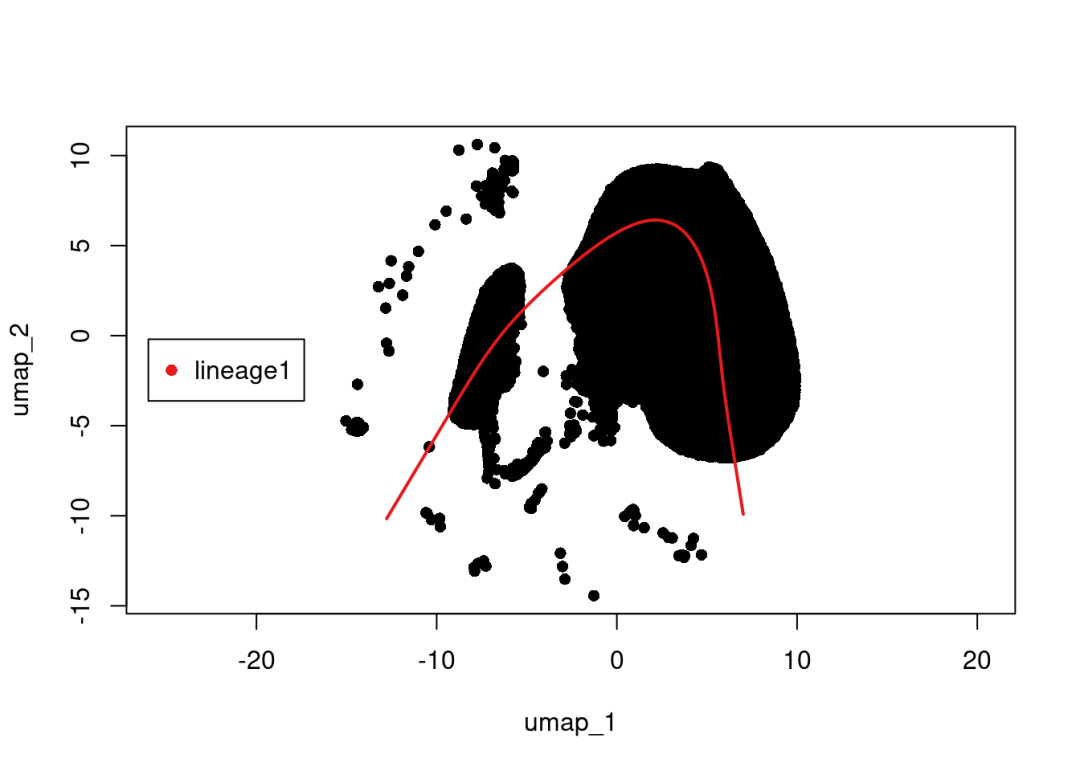
由于monocle2无法分析超过4.5w个细胞类型(可参考5w单细胞在Monocle2拟时序中占据多少内存?)。这里我们以slingshot为例进行演示,详细教程与解读可见Slingshot拟时序分析学习手册
### 加载包 ###
if(!require(Seurat))install.packages("Seurat")
if(!require(dplyr))install.packages("dplyr")
if(!require(ggplot2))install.packages("ggplot2")
if(!require(scran))install.packages("scran")
if(!require(patchwork))install.packages("patchwork")
if(!require(tidyr))install.packages("tidyr")
if(!require(scater))install.packages("scater")
if(!require(stringr))install.packages("stringr")
if(!require(tibble))install.packages("tibble")
if(!require(SingleCellExperiment))install.packages("SingleCellExperiment")
library(hdf5r)
library(Matrix)
library(openxlsx)
library(clustree)
library(paletteer)
library(slingshot)
peakRAM({
scRNA <-readRDS('data/scRNA.rds')
Idents(scRNA) <-'celltype'
cluster_list<-unique(scRNA$celltype)
table(scRNA$celltype)
# 显然大家不可能把所有细胞放在一起做拟时序分析,所以这里我们用最多的两个细胞类型之间进行拟时序分析
select_cluster =as.SingleCellExperiment(subset(scRNA, idents =c('PCT','TAL')), assay ='RNA')
sim <-slingshot(select_cluster,
clusterLabels ='celltype',
reducedDim ='UMAP',
start.clus="PEC",
end.clus ="PCT"
)
SlingshotDataSet(sim)
plot(reducedDims(sim)$UMAP, pch=16, asp =1)
lines(SlingshotDataSet(sim), lwd=2, col=brewer.pal(9,"Set1"))
legend("right",
legend =paste0("lineage",1),
col =unique(brewer.pal(6,"Set1")),
inset=0.8,
pch =16)
# 后面的可视化就不演示啦,感兴趣的小伙伴参考:https://mp.weixin.qq.com/s/eIcjE3Na2EM86zR_I5efnA?payreadticket=HAk1yhmH_rFch77p2LQSC514-riJ6juSFBX1imWY-UCTRd2UNMS4ykhPWaMuEPYYlE3Moy0
})
## Function_Call
## 1 {scRNA<-readRDS("data/scRNA.rds")Idents(scRNA)<-"celltype"cluster_list<-unique(scRNA$celltype)table(scRNA$celltype)select_cluster=as.SingleCellExperiment(subset(scRNA,idents=c("PCT","TAL")),assay="RNA")sim<-slingshot(select_cluster,clusterLabels="celltype",reducedDim="UMAP",start.clus="PEC",end.clus="PCT")SlingshotDataSet(sim)plot(reducedDims(sim)$UMAP,pch=16,asp=1)lines(SlingshotDataSet(sim),lwd=2,col=brewer.pal(9,"Set1"))legend("right",legend=paste0("lineage",1),col=unique(brewer.pal(6,"Set1")),inset=0.8,pch=16)}
## Elapsed_Time_sec Total_RAM_Used_MiB Peak_RAM_Used_MiB
## 1 7263.776 58389.8 95028.6
rm(list =ls());gc()
## used (Mb) gc trigger (Mb) max used (Mb)
## Ncells 15353016 820.0 34294643 1831.6 34294643 1831.6
## Vcells 32342232 246.8 12186755452 92977.6 12485370569 95255.95、 去批次
公认的单细胞中最耗费内存和时间的去批次方法CCA,咱来试一下
peakRAM({
library(Seurat)
library(future)
# 顺序计算,并行计算内存遭不住
plan(strategy ='sequential')
scRNA <-readRDS('data/scRNA.rds')
scRNA_list <-SplitObject(scRNA, split.by ="group")
# 节省点内存,这会scRNA对象可以环境中清除了:
rm(scRNA);gc()
sc.anchors <-FindIntegrationAnchors(object.list = scRNA_list, dims =1:20)# 寻找数据锚定点
scRNA.inte <-IntegrateData(anchorset = sc.anchors, dims =1:20)# 根据锚定点整合数据
scRNA.inte
saveRDS(scRNA.inte,'data/scRNA.inte.rds')
})
## Function_Call
## 1 {library(Seurat)library(future)plan(strategy="sequential")scRNA<-readRDS("data/scRNA.rds")scRNA_list<-SplitObject(scRNA,split.by="group")rm(scRNA)gc()sc.anchors<-FindIntegrationAnchors(object.list=scRNA_list,dims=1:20)scRNA.inte<-IntegrateData(anchorset=sc.anchors,dims=1:20)scRNA.intesaveRDS(scRNA.inte,"data/scRNA.inte.rds")}
## Elapsed_Time_sec Total_RAM_Used_MiB Peak_RAM_Used_MiB
## 1 690265.4 109497.4 383677.3
# 记录一下运行后的时间
Sys.time()
## [1] "2025-09-08 14:45:30 UTC"三、总结
可以看出,我们这次测试还是下了血本的。就计算结果而言,最占内存、时间的任然是大家永远的噩梦------CCA,共花费了374.7GB的内存(仅仅是单线程顺序计算哦)与191.74h的时间。我们可以进行如下可视化来直观的观察:
library(ggplot2)
library(tidyr)
# 数据
data <- data.frame(
Step = factor(c("基础分析", "细胞通讯", "拟时序分析", "去批次(CCA)"),
levels = c("基础分析", "细胞通讯", "拟时序分析", "去批次(CCA)")),
Memory_GB = c(120.4, 123.0, 92.8, 374.7),
Time_Hours = c(4.14, 1.04, 2.02, 191.74)
)
# 内存占用可视化
p1 <- ggplot(data, aes(x = Step, y = Memory_GB, fill = Step)) +
geom_bar(stat = "identity") +
labs(title = "各分析步骤内存占用 (GB)", y = "内存 (GB)", x = "") +
theme_minimal() +
theme(axis.text.x = element_text(angle = 45, hjust = 1)) +
scale_fill_brewer(palette = "Set2")
# 计算时间可视化
p2 <- ggplot(data, aes(x = Step, y = Time_Hours, fill = Step)) +
geom_bar(stat = "identity") +
labs(title = "各分析步骤计算时间 (小时)", y = "时间 (小时)", x = "") +
theme_minimal() +
theme(axis.text.x = element_text(angle = 45, hjust = 1)) +
scale_fill_brewer(palette = "Set3")
# 并排显示
library(patchwork)
p3 <- p1 + p2 + plot_layout(ncol = 2)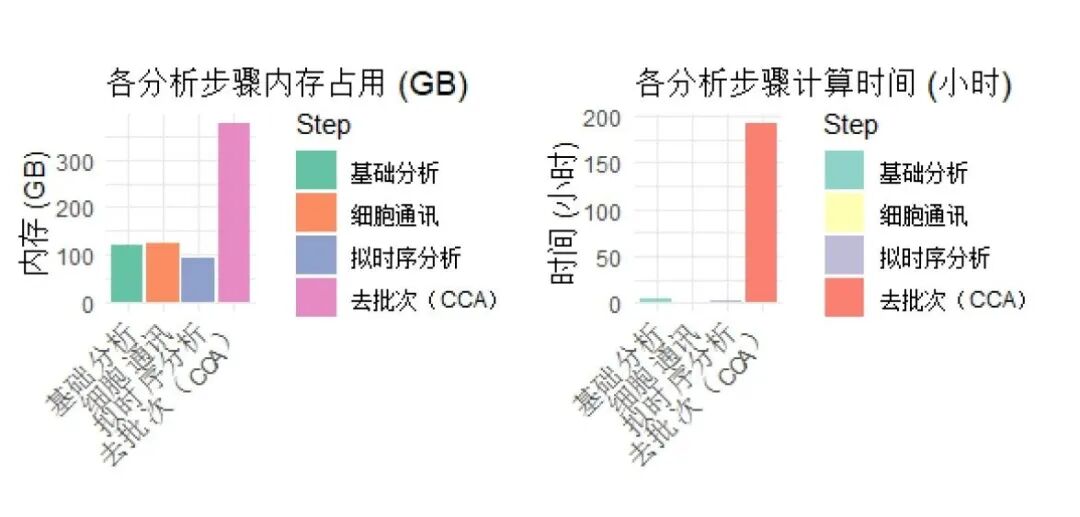
四、演示环境
一切不提供计算环境信息的教程都是耍流氓:
sessionInfo()
## R version 4.4.2 (2024-10-31)
## Platform: x86_64-pc-linux-gnu
## Running under: Ubuntu 20.04.4 LTS
##
## Matrix products: default
## BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
## LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/liblapack.so.3; LAPACK version 3.9.0
##
## locale:
## [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
## [3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
## [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
## [7] LC_PAPER=en_US.UTF-8 LC_NAME=C
## [9] LC_ADDRESS=C LC_TELEPHONE=C
## [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
##
## time zone: Etc/UTC
## tzcode source: system (glibc)
##
## attached base packages:
## [1] stats4 stats graphics grDevices utils datasets methods
## [8] base
##
## other attached packages:
## [1] slingshot_2.14.0 TrajectoryUtils_1.14.0
## [3] princurve_2.1.6 paletteer_1.6.0
## [5] clustree_0.5.1 ggraph_2.2.1
## [7] openxlsx_4.2.8 Matrix_1.7-2
## [9] hdf5r_1.3.12 tibble_3.2.1
## [11] stringr_1.5.1 scater_1.34.0
## [13] tidyr_1.3.1 scran_1.30.2
## [15] scuttle_1.12.0 SingleCellExperiment_1.28.1
## [17] SummarizedExperiment_1.36.0 GenomicRanges_1.58.0
## [19] GenomeInfoDb_1.42.1 IRanges_2.40.1
## [21] S4Vectors_0.44.0 MatrixGenerics_1.18.1
## [23] matrixStats_1.5.0 ggalluvial_0.12.5
## [25] future_1.34.0 biovizBase_1.54.0
## [27] colorspace_2.1-1 RColorBrewer_1.1-3
## [29] patchwork_1.3.0 peakRAM_1.0.2
## [31] CellChat_2.1.2 bigmemory_4.6.4
## [33] Biobase_2.66.0 BiocGenerics_0.52.0
## [35] ggplot2_3.5.1 igraph_2.1.4
## [37] dplyr_1.1.4 Seurat_5.2.1
## [39] SeuratObject_5.0.2 sp_2.2-0
##
## loaded via a namespace (and not attached):
## [1] R.methodsS3_1.8.2 dichromat_2.0-0.1
## [3] airports_0.1.0 nnet_7.3-20
## [5] goftest_1.2-3 Biostrings_2.74.1
## [7] vctrs_0.6.5 spatstat.random_3.4-1
## [9] digest_0.6.37 png_0.1-8
## [11] shape_1.4.6.1 registry_0.5-1
## [13] ggrepel_0.9.6 deldir_2.0-4
## [15] parallelly_1.42.0 MASS_7.3-64
## [17] reshape2_1.4.4 httpuv_1.6.15
## [19] foreach_1.5.2 withr_3.0.2
## [21] cherryblossom_0.1.0 xfun_0.50
## [23] ggpubr_0.6.0 survival_3.8-3
## [25] memoise_2.0.1 ggbeeswarm_0.7.2
## [27] systemfonts_1.2.1 zoo_1.8-12
## [29] GlobalOptions_0.1.2 pbapply_1.7-2
## [31] R.oo_1.27.0 Formula_1.2-5
## [33] usdata_0.3.1 rematch2_2.1.2
## [35] KEGGREST_1.46.0 promises_1.3.2
## [37] httr_1.4.7 rstatix_0.7.2
## [39] restfulr_0.0.15 globals_0.16.3
## [41] fitdistrplus_1.2-2 rstudioapi_0.17.1
## [43] UCSC.utils_1.2.0 miniUI_0.1.1.1
## [45] generics_0.1.3 base64enc_0.1-3
## [47] curl_6.2.0 zlibbioc_1.52.0
## [49] ScaledMatrix_1.14.0 polyclip_1.10-7
## [51] GenomeInfoDbData_1.2.13 SparseArray_1.6.0
## [53] xtable_1.8-4 doParallel_1.0.17
## [55] evaluate_1.0.3 S4Arrays_1.6.0
## [57] hms_1.1.3 irlba_2.3.5.1
## [59] ggnetwork_0.5.13 ROCR_1.0-11
## [61] reticulate_1.43.0.9001 spatstat.data_3.1-6
## [63] magrittr_2.0.3 lmtest_0.9-40
## [65] readr_2.1.5 viridis_0.6.5
## [67] later_1.4.1 lattice_0.22-6
## [69] spatstat.geom_3.4-1 NMF_0.28
## [71] future.apply_1.11.3 scattermore_1.2
## [73] XML_3.99-0.18 cowplot_1.1.3
## [75] RcppAnnoy_0.0.22 Hmisc_5.2-2
## [77] pillar_1.10.1 nlme_3.1-168
## [79] iterators_1.0.14 sna_2.8
## [81] gridBase_0.4-7 compiler_4.4.2
## [83] beachmat_2.22.0 RSpectra_0.16-2
## [85] stringi_1.8.4 tensor_1.5
## [87] GenomicAlignments_1.42.0 plyr_1.8.9
## [89] crayon_1.5.3 abind_1.4-8
## [91] BiocIO_1.16.0 locfit_1.5-9.11
## [93] graphlayouts_1.2.2 bit_4.5.0.1
## [95] codetools_0.2-20 BiocSingular_1.22.0
## [97] bslib_0.9.0 GetoptLong_1.0.5
## [99] plotly_4.10.4 mime_0.12
## [101] splines_4.4.2 circlize_0.4.16
## [103] Rcpp_1.0.14 fastDummies_1.7.5
## [105] sparseMatrixStats_1.18.0 knitr_1.49
## [107] blob_1.2.4 clue_0.3-66
## [109] AnnotationFilter_1.30.0 listenv_0.9.1
## [111] checkmate_2.3.2 DelayedMatrixStats_1.28.1
## [113] ggsignif_0.6.4 statmod_1.5.0
## [115] tzdb_0.4.0 svglite_2.1.3
## [117] tweenr_2.0.3 pkgconfig_2.0.3
## [119] network_1.19.0 tools_4.4.2
## [121] cachem_1.1.0 RSQLite_2.3.9
## [123] viridisLite_0.4.2 DBI_1.2.3
## [125] fastmap_1.2.0 rmarkdown_2.29
## [127] scales_1.3.0 grid_4.4.2
## [129] ica_1.0-3 Rsamtools_2.22.0
## [131] broom_1.0.7 sass_0.4.9
## [133] coda_0.19-4.1 FNN_1.1.4.1
## [135] BiocManager_1.30.25 dotCall64_1.2
## [137] VariantAnnotation_1.52.0 carData_3.0-5
## [139] RANN_2.6.2 rpart_4.1.24
## [141] farver_2.1.2 tidygraph_1.3.1
## [143] yaml_2.3.10 foreign_0.8-88
## [145] rtracklayer_1.66.0 cli_3.6.3
## [147] purrr_1.0.2 lifecycle_1.0.4
## [149] uwot_0.2.2 presto_1.0.0
## [151] bluster_1.12.0 backports_1.5.0
## [153] BiocParallel_1.40.0 gtable_0.3.6
## [155] rjson_0.2.23 ggridges_0.5.6
## [157] progressr_0.15.1 parallel_4.4.2
## [159] limma_3.62.2 jsonlite_1.8.9
## [161] edgeR_4.4.1 RcppHNSW_0.6.0
## [163] bitops_1.0-9 bigmemory.sri_0.1.8
## [165] bit64_4.6.0-1 Rtsne_0.17
## [167] openintro_2.5.0 spatstat.utils_3.1-4
## [169] BiocNeighbors_1.20.2 zip_2.3.1
## [171] jquerylib_0.1.4 metapod_1.14.0
## [173] dqrng_0.4.1 spatstat.univar_3.1-3
## [175] R.utils_2.12.3 lazyeval_0.2.2
## [177] shiny_1.10.0 htmltools_0.5.8.1
## [179] sctransform_0.4.1 ensembldb_2.30.0
## [181] glue_1.8.0 spam_2.11-1
## [183] XVector_0.46.0 RCurl_1.98-1.16
## [185] BSgenome_1.74.0 gridExtra_2.3
## [187] R6_2.5.1 GenomicFeatures_1.58.0
## [189] cluster_2.1.8 rngtools_1.5.2
## [191] statnet.common_4.11.0 vipor_0.4.7
## [193] DelayedArray_0.32.0 tidyselect_1.2.1
## [195] ProtGenerics_1.38.0 htmlTable_2.4.3
## [197] ggforce_0.4.2 car_3.1-3
## [199] AnnotationDbi_1.68.0 rsvd_1.0.5
## [201] munsell_0.5.1 KernSmooth_2.23-26
## [203] data.table_1.16.4 htmlwidgets_1.6.4
## [205] ComplexHeatmap_2.12.1 rlang_1.1.5
## [207] spatstat.sparse_3.1-0 spatstat.explore_3.4-3
## [209] uuid_1.2-1 beeswarm_0.4.0