生信碱移
快速CNV分析
fastCNV 是scRNA-seq与空间转录组(含Visium HD)的快速 CNV 推断工具,比常规工具 inferCNV 运行速度快 10 倍,平均内存占用减少 94%。不仅如此,fastCNV 通过将 spots/cells 聚合为 meta-spots/meta-cells 以增强信号,输出全基因组 CNV 热图,并进行 CNV 聚类、克隆树推断与染色体臂级别 CNV 事件注释。
DNA拷贝数变异(CNV)在肿瘤发生发展中占据核心地位,其既可能通过抑癌基因缺失或致癌基因扩增驱动病理过程,也会在肿瘤演化中逐步累积,塑造显著的肿瘤内异质性,并影响生长、耐药与预后。
临床上,CNV 通常依赖 WES/WGS 等 DNA 测序手段进行检测。单细胞转录组与空间转录组(ST)在解析组织结构与细胞状态方面优势突出,如果能够从这些转录组数据中推断CNV,就有机会以更低成本、更高空间/细胞分辨率描绘肿瘤克隆结构。尽管如此,现有 CNV 分析工具难以处理大规模数据,包括运行速度慢、内存消耗高等。
小编也是苦运行速度久矣,这不最近冲浪的时候发现了一个刚刚上线的 R 包 fastCNV,能够实现面向 scRNA-seq 与空间转录组的快速精确 CNV 推断。
一开始不得不提一下 fastCNV 的三大特点,具体带大家根据原文的分析结果来看看。
特点一:更高的 CNV 推断准确性。作者选择 117 个 CCLE 癌细胞系,这些细胞系具有全基因组的配对结果。总体来说,fastCNV 与 WES 的相关性整体较高,raw 分数的中位相关性为 0.76(均值0.72),denoised 分数中位数为 0.71(均值0.67)。对比 inferCNV 时,fastCNV 无论在 raw 还是 denoised 层面都显著优于 inferCNV(配对T检验p值分别小于4e-31与8e-24)。
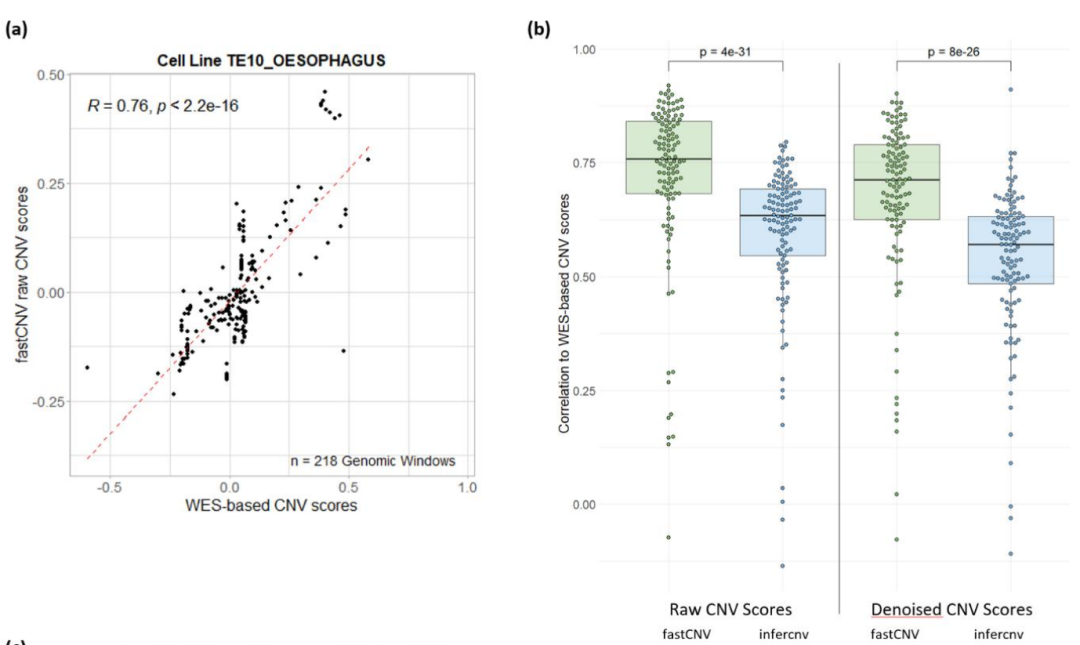
图:fastCNV 与 inferCNV 的性能比较。作者分别对每个细胞系的 scRNA-seq 数据分别运行 fastCNV 与 inferCNV,最终与 WES 推测的 CNV 在同一基因组区域尺度上计算相关性。a图显示 fastCNV 具有高相关性,b图显示 fastCNV 由于inferCNV。
特点二:快速且更低内存占用的 CNV 计算。具体来说,fastCNV 在 117 个细胞系上完成分析需要约 4 分钟,而 inferCNV 则需要约 40 分钟(运行时间快了 10 倍啊)。在此基础上,其平均内存仅为 80 MiB(内存占用约为 inferCNV 的6%),峰值内存约 19 GiB(inferCNV 约 32 GiB)。
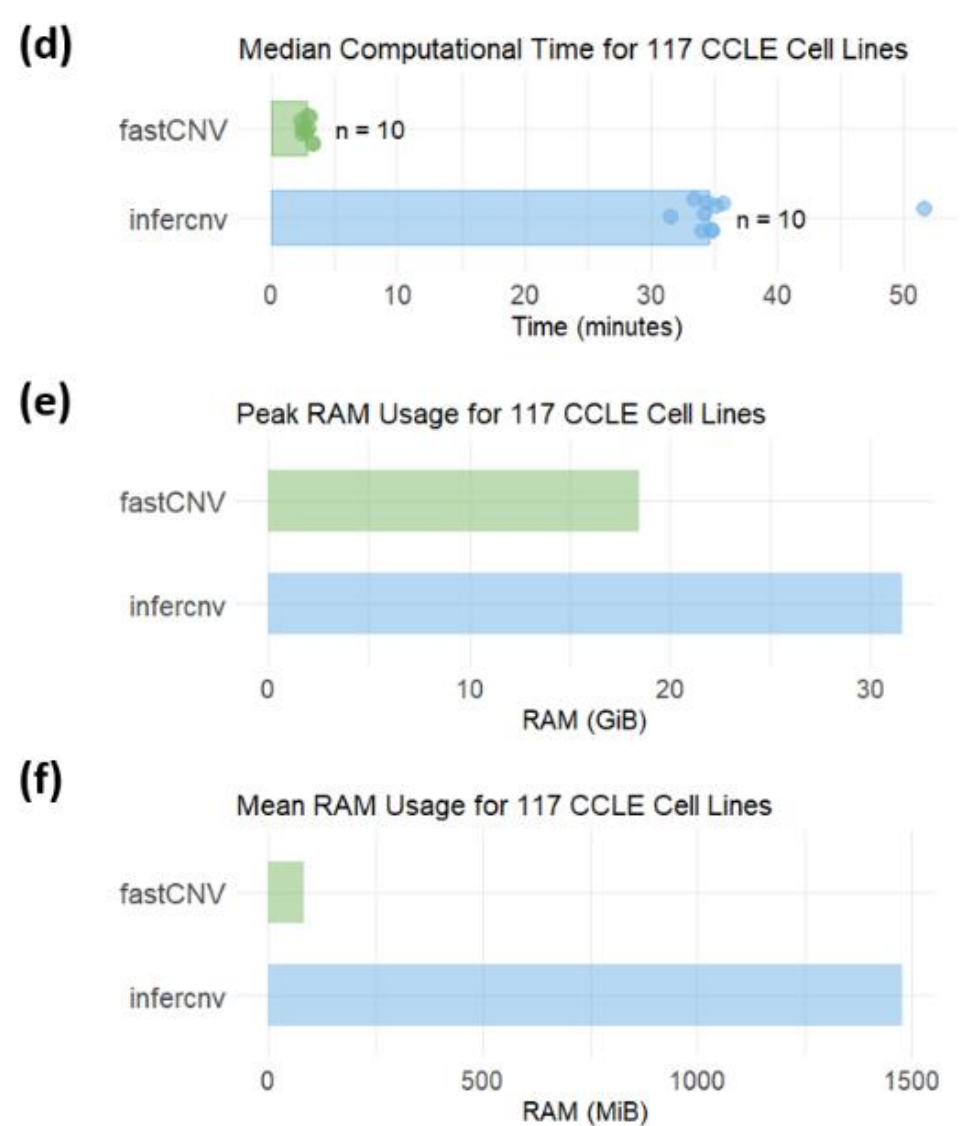
图:10次运行下 fastCNV 与 inferCNV 的时间/内存占用比较。
特点三:空间转录组的 CNV 推断与空间亚克隆/演化分析功能。fastCNV 不仅在 scRNA-seq 上做 CNV 打分和聚类,还能把 CNV 簇映射回组织空间位置,用 CNV fraction 等指标区分肿瘤与非肿瘤区域。
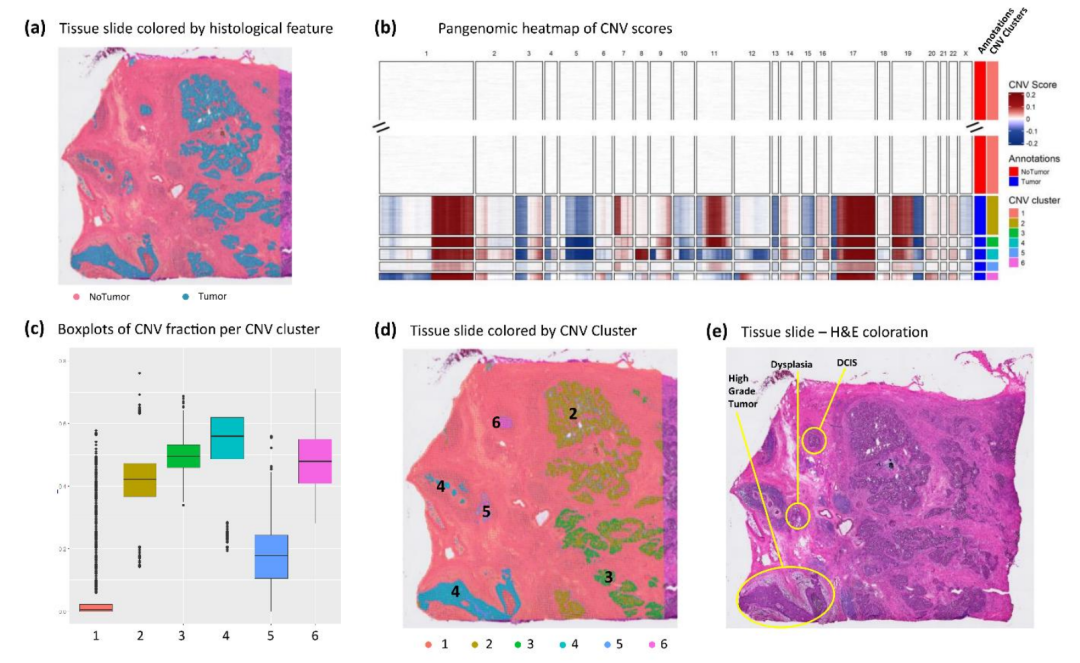
图:基于CNV的聚类与打分。
在此基础上能够进一步重建空间层面的克隆结构与克隆树,探究亚克隆出现以及肿瘤演化顺序。
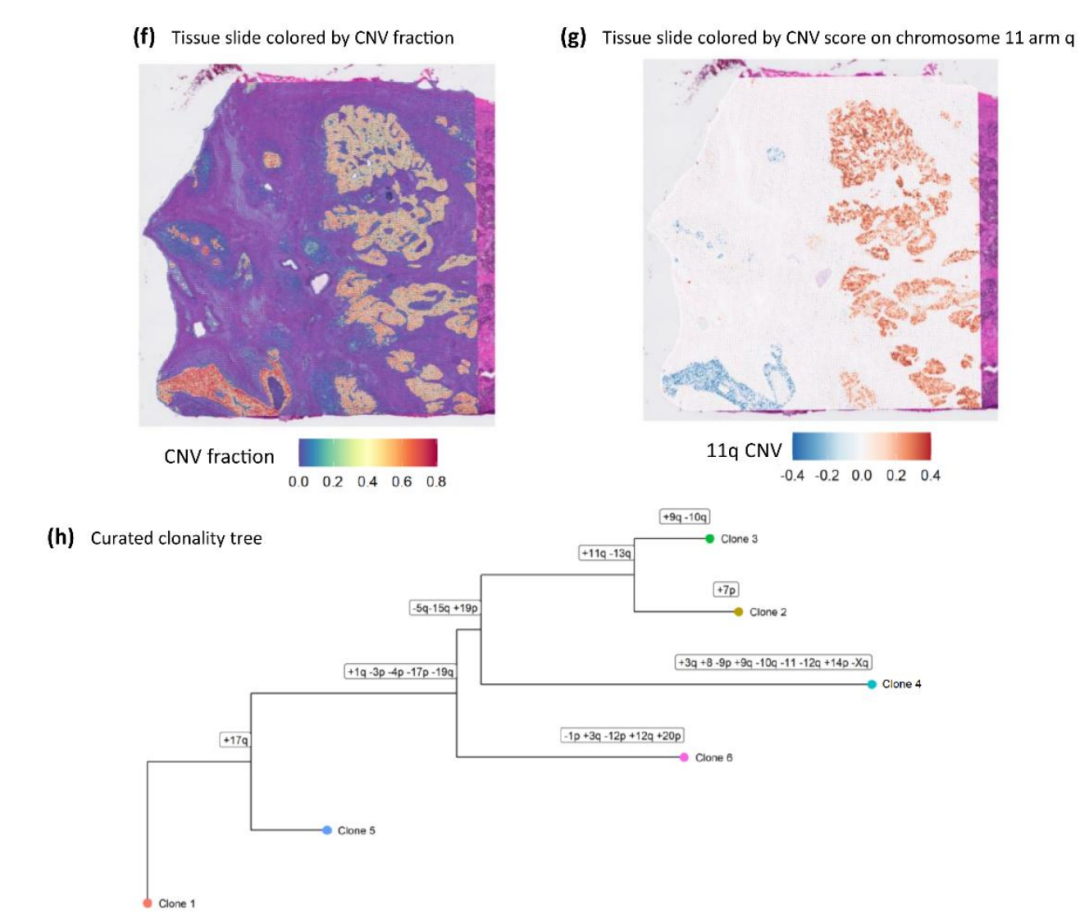
图:CNV得分与亚克隆树构建。
fastCNV 的使用和原理层面且听小编婉婉道来。简单来说,其以 Seurat对象(单样本或多样本列表)作为输入,输出包含 CNV 结果的 Seurat 对象,并可生成类似于 inferCNV 全基因组热图。具体的流程如下:① fastCNV 先用 Louvain 聚类并簇内聚合生成meta cell以增强低读数信号,② 继而基于 Ensembl 将高表达基因按染色体臂滑窗划分区域,③ 对 log2 零中心化表达以二倍体参考校正得到 raw 并截断;④ 再按参考分位去噪生成 denoised 与 CNV fraction,⑤ 最后以层次聚类确定亚克隆并由 CNVTree 基于簇中心构建克隆树并标注臂级增益/缺失。
本文简要介绍 fastCNV 在单细胞与空转数据上的使用,感兴趣的铁子可以进一步阅读原文与 github:
-
github链接: https://github.com/must-bioinfo/fastCNV
-
原文DOI:10.1101/2025.10.22.683855
0.R包安装
fastCNV 仅适用于 Seurat5 版本,具体可以使用以下代码安装该包:
# R包
remotes::install_github("must-bioinfo/fastCNV")
# 示例数据
remotes::install_github("must-bioinfo/fastCNVdata")1.单细胞fastCNV分析
① 加载示例数据:
library(fastCNV)
library(fastCNVdata)
# 加载 scColon1、scColon2、scColon3、scColon4
# 为结直肠肿瘤单细胞测序数据
# 具体信息见作者原文
utils::data(scColon1)
# 查看数据注释
unique(scColon1[["annot"]])② 执行fastCNV分析:
scColon1 <- fastCNV(seuratObj = scColon1,
sampleName = "scColon1",
referenceVar = "annot",
referenceLabel = c("TNKILC", "Myeloid", "B","Mast", "Plasma"),
printPlot = T)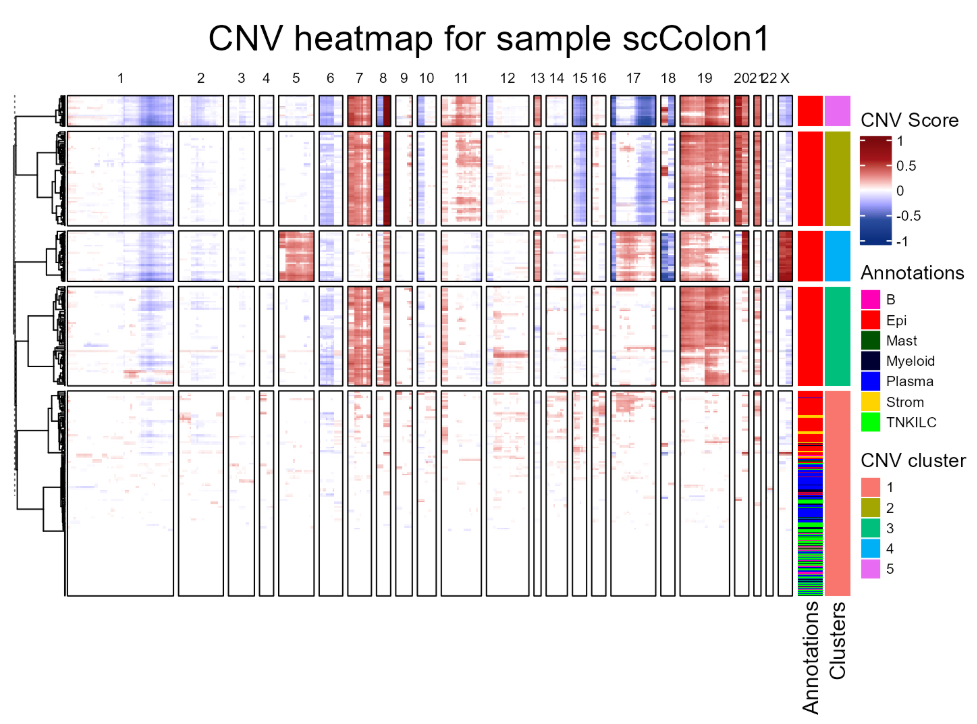
上述代码在scColon1对象上运行fastCNV()进行CNV推断,其中以annot列中标注为 TNKILC、Myeloid、B、Mast、Plasma 的细胞作为参考二倍体群体(拷贝数正常的细胞群体),并将样本命名为scColon1且输出结果图(printPlot=TRUE)。
③ cnv得分计算。fastCNV 会为 Seurat 对象中的每个观测值计算一个 cnv_fraction (CNV 得分)。可以直接使用 Seurat 的绘图函数对其进行绘制:
library(Seurat)
library(ggplot2)
scColon1 <- RunUMAP(scColon1, dims = 1:10)
common_theme <- theme(
plot.title = element_text(size = 10),
legend.text = element_text(size = 8),
legend.title = element_text(size = 8),
axis.title = element_text(size = 8),
axis.text = element_text(size = 6)
)
FeaturePlot(scColon1, features = "cnv_fraction", reduction = "umap" ) & common_theme |
DimPlot(scColon1, reduction = "umap", group.by = "annot") & common_theme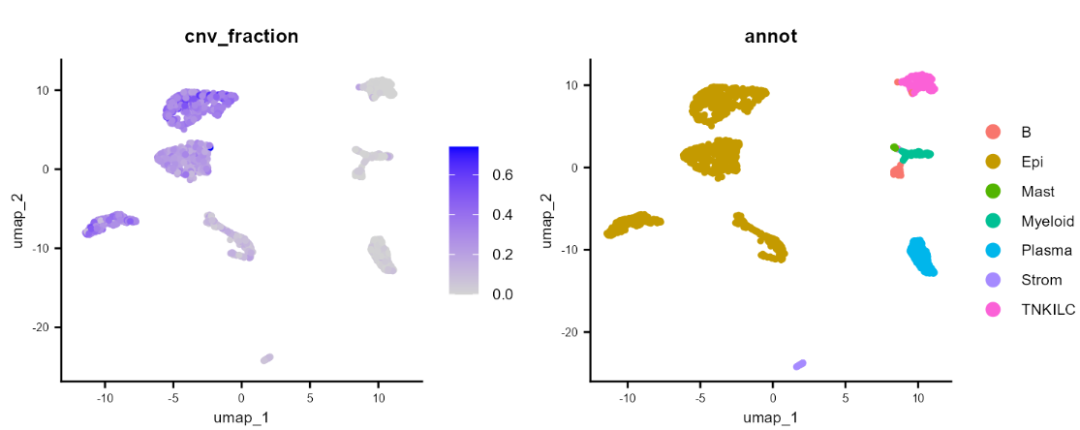
画个boxplot,可以看到一些细胞亚群的拷贝数变异(CNV)分数比其他簇高得多:
ggplot(FetchData(scColon1, vars = c("annot", "cnv_fraction")),
aes(annot, cnv_fraction, fill = annot)) +
geom_boxplot() +
theme(axis.text.x = element_text(angle = 45, vjust = 1, hjust = 1, color = "black"))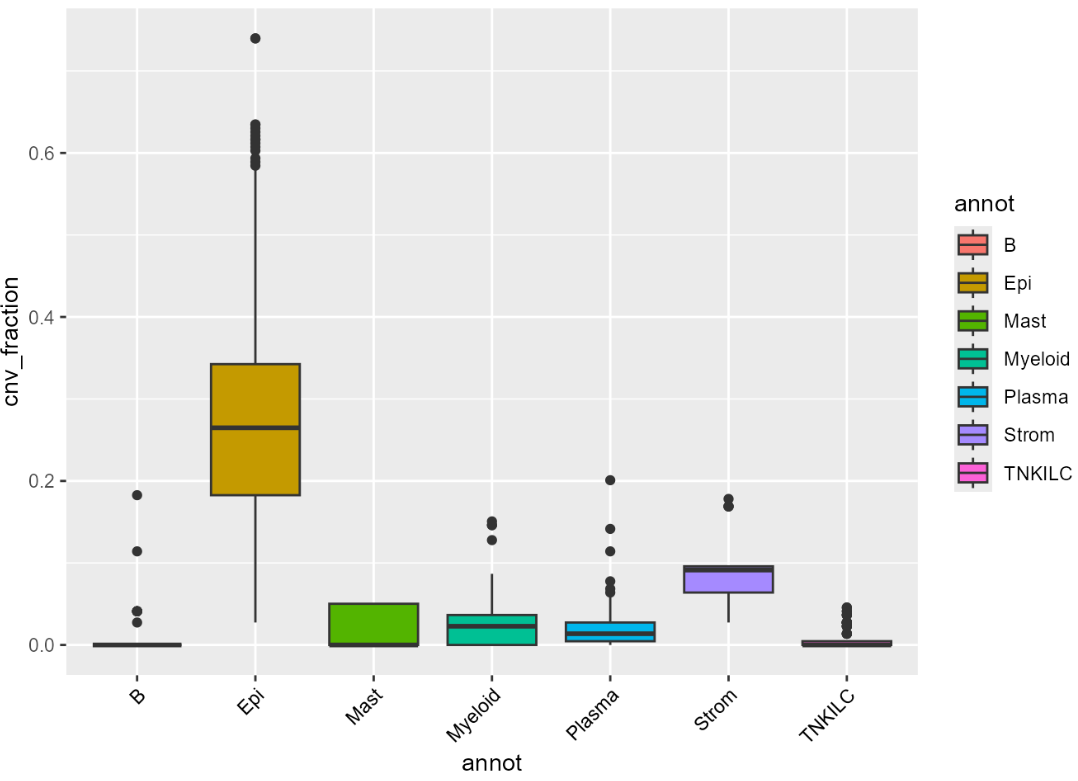
在此基础上,还可以使用 Seurat 的绘图函数来绘制按染色体臂划分的 CNV。对于某些肿瘤,其在特定染色体中会存在显著 CNV 变化的:
library(scales)
FeaturePlot(scColon1, features = "20.p_CNV") +
scale_color_distiller(palette = "RdBu", direction = -1, limits = c(-1, 1),
rescaler = function(x, to = c(0, 1), from = NULL) {
rescale_mid(x, to = to, mid = 0)
}) +
common_theme |
FeaturePlot(scColon1, features = "X.q_CNV") +
scale_color_distiller(palette = "RdBu", direction = -1, limits = c(-1, 1),
rescaler = function(x, to = c(0, 1), from = NULL) {
rescale_mid(x, to = to, mid = 0)
}) +
common_theme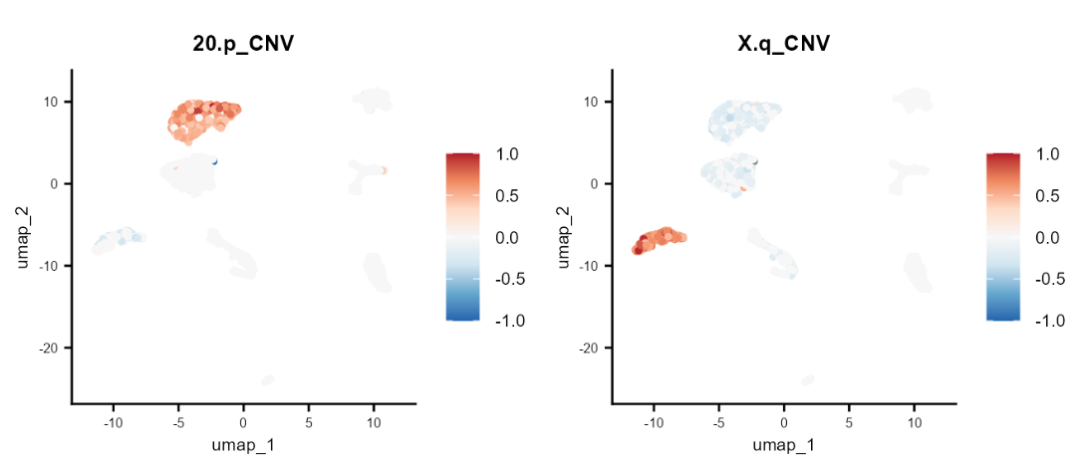
④ CNV 分类。通过使用按染色体臂划分的 CNV 和 CNVclassification函数,可以获得根据染色体臂改变的分类:
scColon1 <- CNVClassification(scColon1)
DimPlot(scColon1, group.by = "20.p_CNV_classification") &
scale_color_manual(values = c(gain = "red", no_alteration = "grey", loss = "blue")) &
common_theme |
DimPlot(scColon1, group.by = "X.q_CNV_classification") &
scale_color_manual(values = c(gain = "red", no_alteration = "grey", loss = "blue")) &
common_theme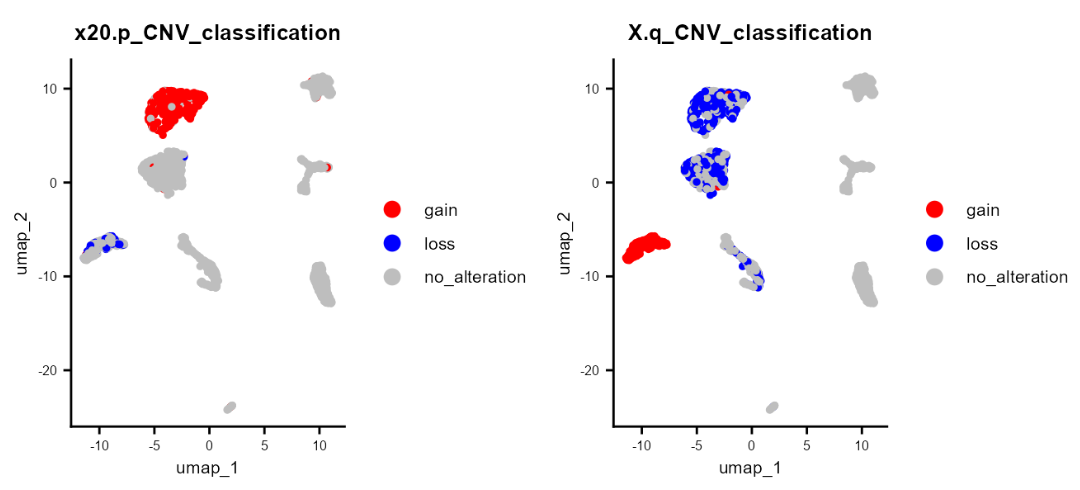
⑤ 基于拷贝数变异的聚类簇。fastCNV 还会计算 CNV 聚类,并将其以 "cnv_clusters" 的形式保存在 Seurat 对象的元数据中:
DimPlot(scColon1, group.by = "cnv_clusters") + common_theme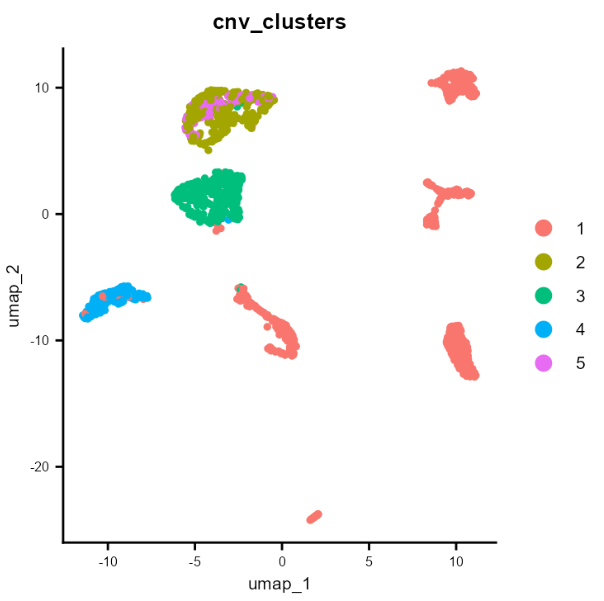
通过这个分析,作者发现肿瘤细胞几乎总是属于2/3/4/5 cnv_cluster;而健康细胞往往是 cnv_cluster 1,几乎没有没有拷贝数变异。
⑥ 亚克隆性树构建。如上一小节所述,scColon1 中有 4 个肿瘤亚簇(2/3/4/5),以及 1 个健康亚簇(1)。在此基础上,可以使用 CNVTree() 来构建亚克隆性树:
tree_data <- CNVTree(scColon1, values = "calls", cnv_thresh = 0.09, healthyClusters = "1")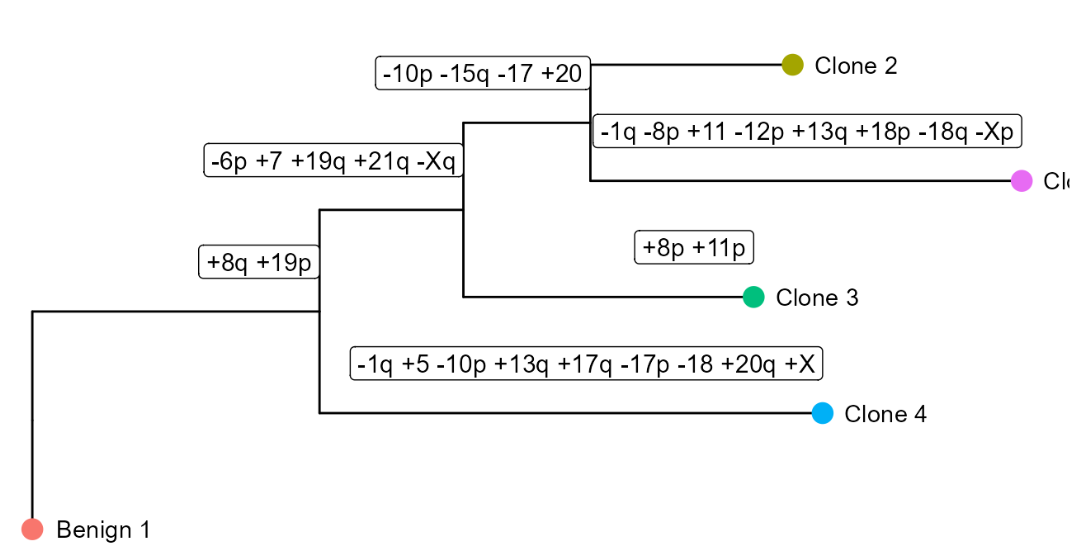
2.空转fastCNV分析
① 示例数据加载 (Visium HD 数据,分辨率更高需要更高的内存占用):
library(fastCNV)
library(fastCNVdata)
library(Seurat)
# 加载数据
HDBreast <- load_HDBreast()
# 查看注释好的肿瘤/非肿瘤区域
unique(HDBreast[["annots_8um"]])
#> annots_8um
#> s_016um_00107_00066-1
#> s_008um_00269_00526-1 NoTumor
#> s_008um_00260_00253-1 Tumor值得强调的是,fastCNV 默认使用 16um 测序,如果大家的注释是在 8um 分辨率上的,则需要在对 16um 测序运行 fastCNV 之前先运行下述转换:
HDBreast <- annotations8umTo16um(HDBreast, referenceVar = "annots_8um")
#> [1] "New annotation column (new referenceVar) is named : projected_annots_8um."上述代码对 16µm 点分配给 8µm 点的注释,生成了projected_annots_8um新的列。在此基础上,可以打印注释看看对不对:
SpatialDimPlot(HDBreast, group.by = "projected_annots_8um")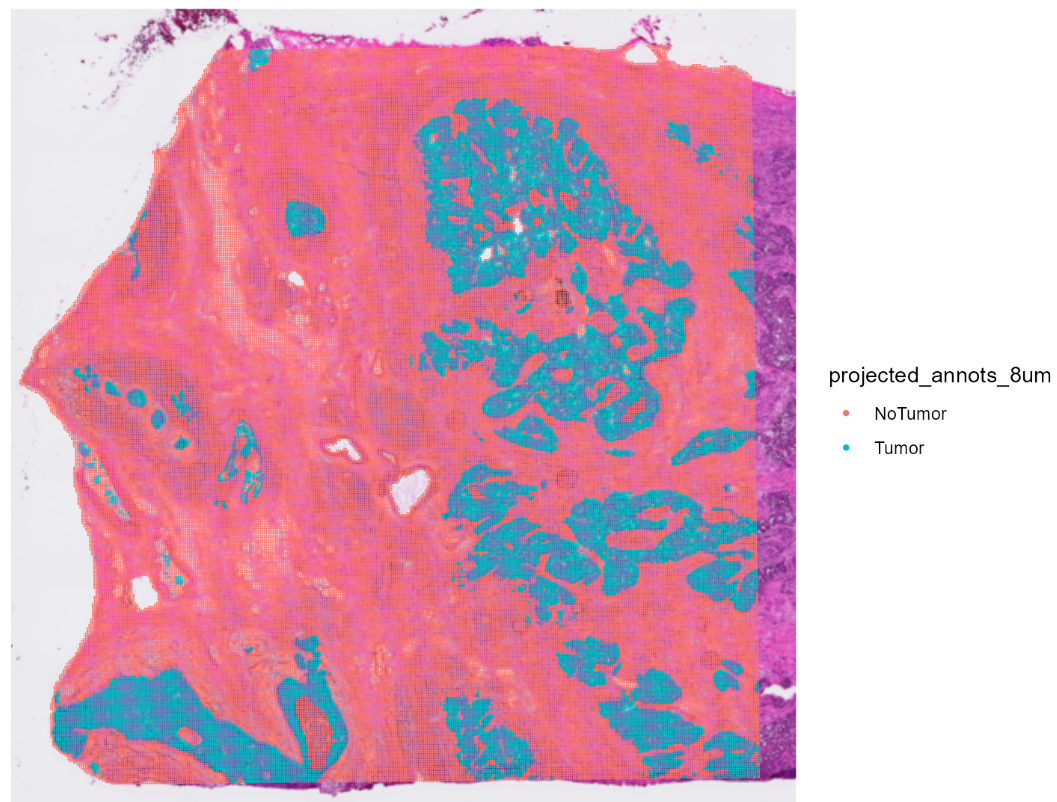
② 运行 fastCNV,参数与单细胞的分析一致。这里将 NoTumor指定为参考:
HDBreast <- fastCNV_10XHD(HDBreast,
sampleName = "HDBreast",
referenceVar = "projected_annots_8um",
referenceLabel = "NoTumor",
printPlot = TRUE)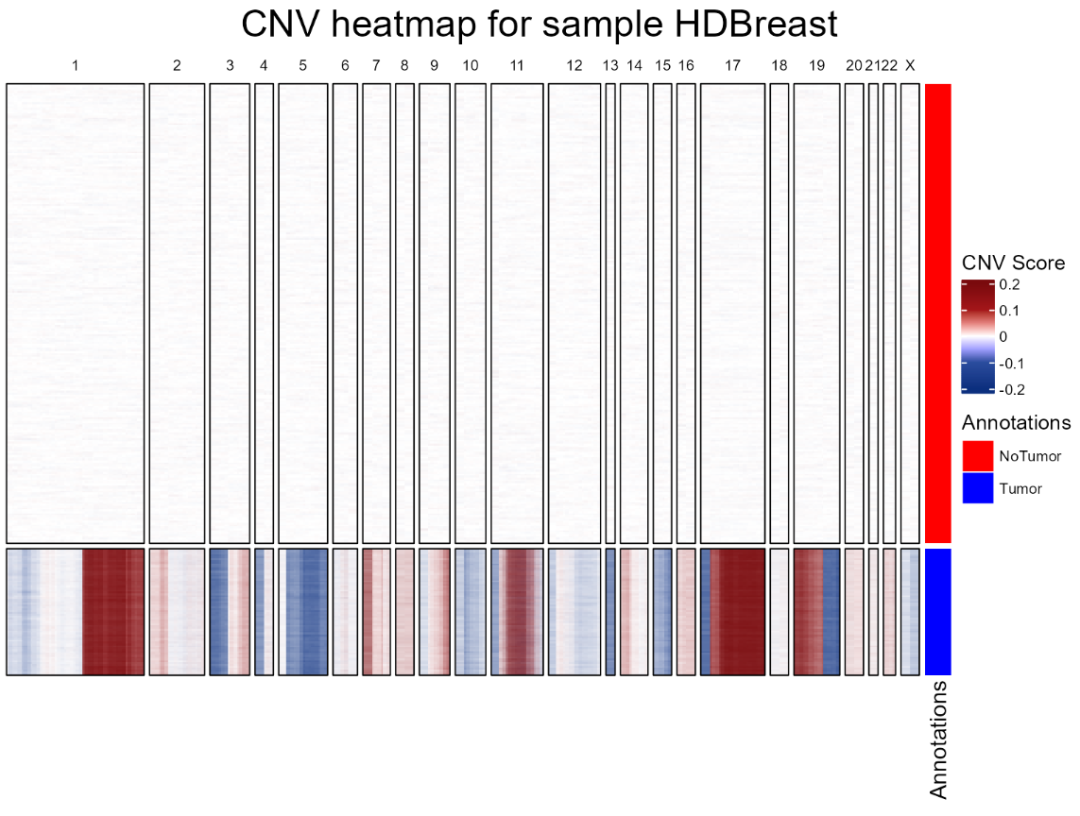
下面的分析就与单细胞类似了,小编不做过多赘述。
③ CNV 分数 (cnv_fraction)。
library(patchwork)
SpatialFeaturePlot(HDBreast, "cnv_fraction") | SpatialPlot(HDBreast, group.by = "projected_annots_8um")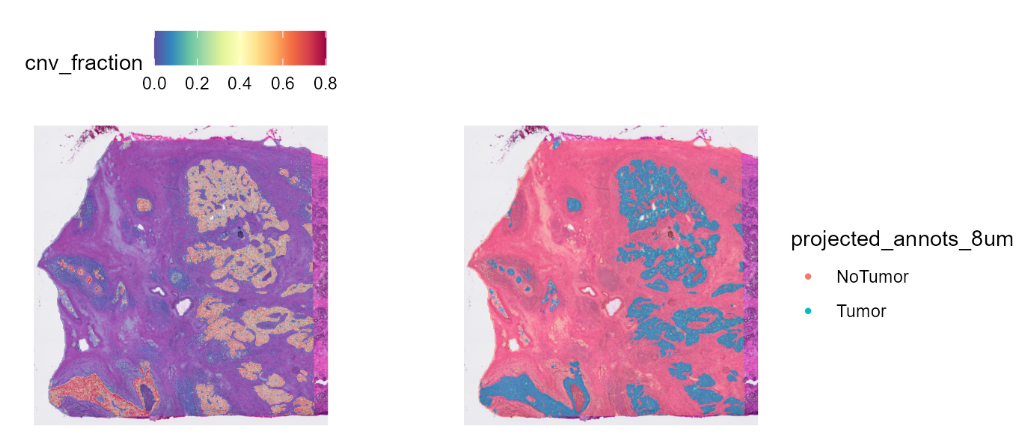
两个区域的CNV差异:
library(ggplot2)
ggplot(FetchData(HDBreast, vars = c("projected_annots_8um", "cnv_fraction")),
aes(projected_annots_8um, cnv_fraction, fill = projected_annots_8um)) +
geom_boxplot() +
theme(axis.text.x = element_text(angle = 45, vjust = 1, hjust = 1, color = "black"))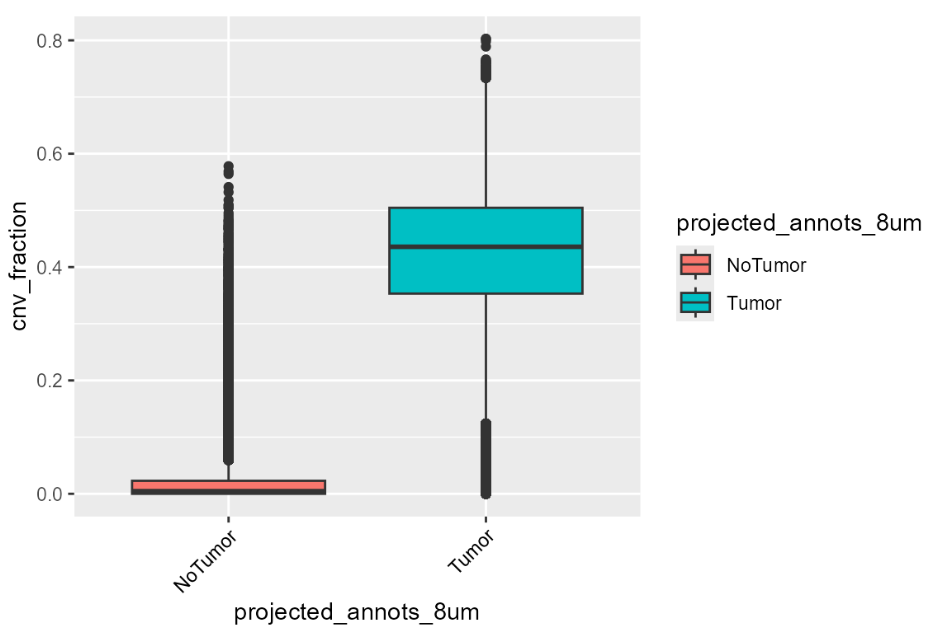
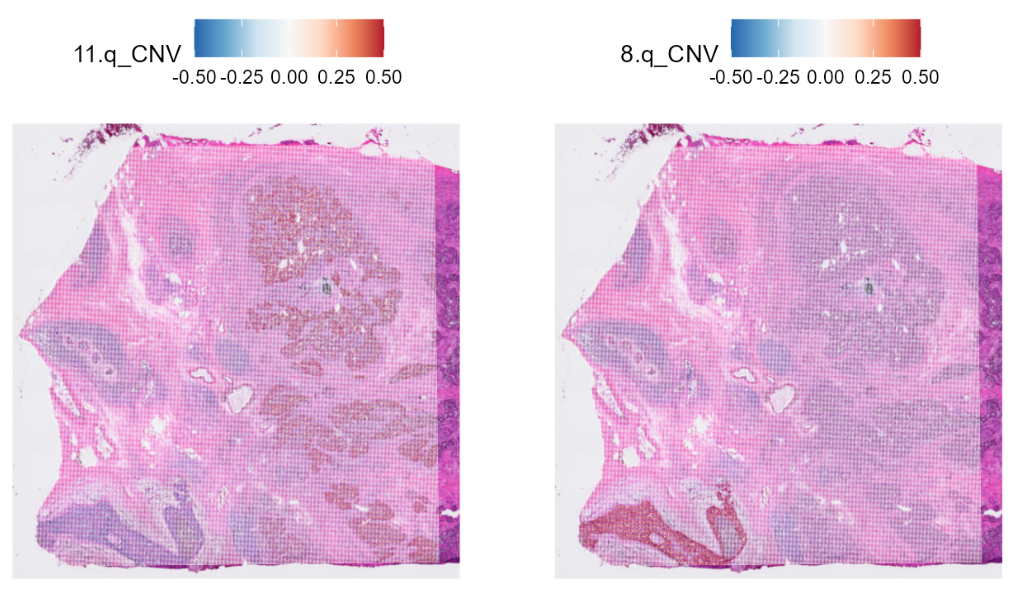
以及按照染色体臂划分的CNV:
library(scales)
SpatialFeaturePlot(HDBreast, features = "11.q_CNV") +
scale_fill_distiller(palette = "RdBu", direction = -1, limits = c(-0.5, 0.5),
rescaler = function(x, to = c(0, 1), from = NULL) {
rescale_mid(x, to = to, mid = 0)
}) |
SpatialFeaturePlot(HDBreast, features = "8.q_CNV") +
scale_fill_distiller(palette = "RdBu", direction = -1, limits = c(-0.5, 0.5),
rescaler = function(x, to = c(0, 1), from = NULL) {
rescale_mid(x, to = to, mid = 0)
})
④ 基于CNV的聚类簇:
HDBreast <- CNVCluster(HDBreast, referenceVar = "projected_annots_8um", tumorLabel = "Tumor")
SpatialDimPlot(HDBreast, group.by = "cnv_clusters") 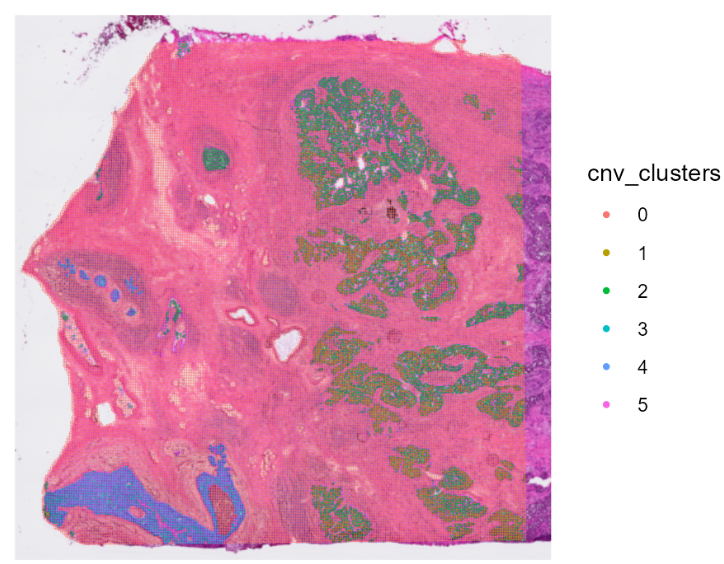
比例展示:
library(ggplot2)
library(SeuratObject)
HDBreast$cnv_clusters <- as.factor(HDBreast$cnv_clusters)
ggplot(FetchData(HDBreast, vars = c("cnv_clusters", "projected_annots_8um")), aes(projected_annots_8um, fill = cnv_clusters)) +
geom_bar(position = "fill") +
theme(axis.text.x = element_text(angle = 45, vjust = 1, hjust = 1, color = "black"))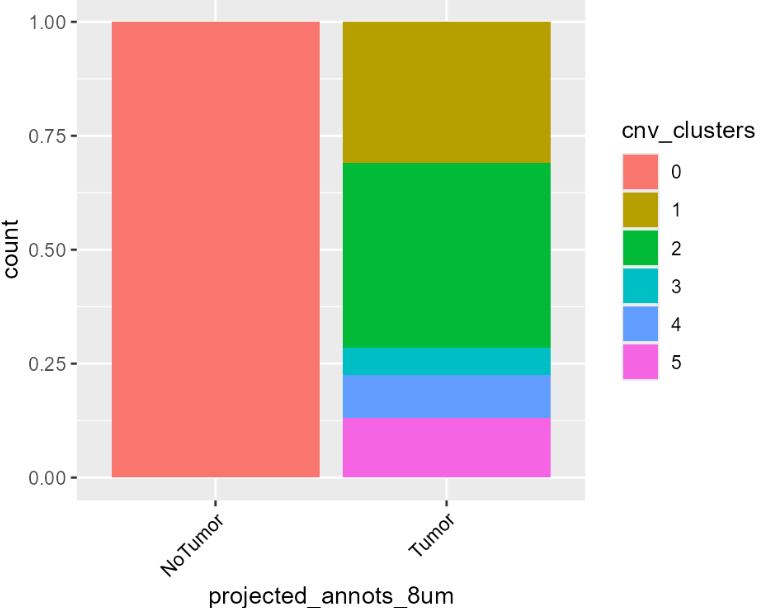
再次绘制 CNV 热图,在热图中可视化这些聚类:
plotCNVResultsHD(HDBreast, referenceVar = "projected_annots_8um", printPlot = TRUE)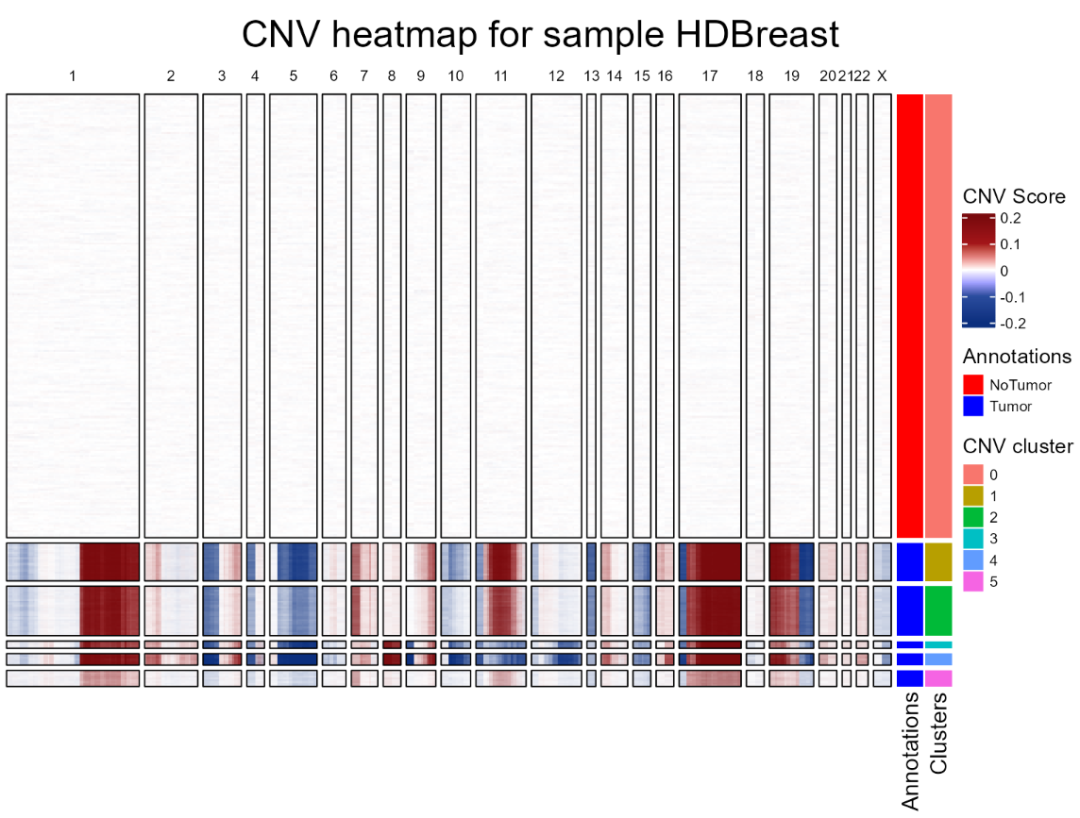
⑤ 构建基于CNV的亚克隆树:
tree_data <- CNVTree(HDBreast, values ="calls", cnv_thresh = 0.09, healthyClusters = "0")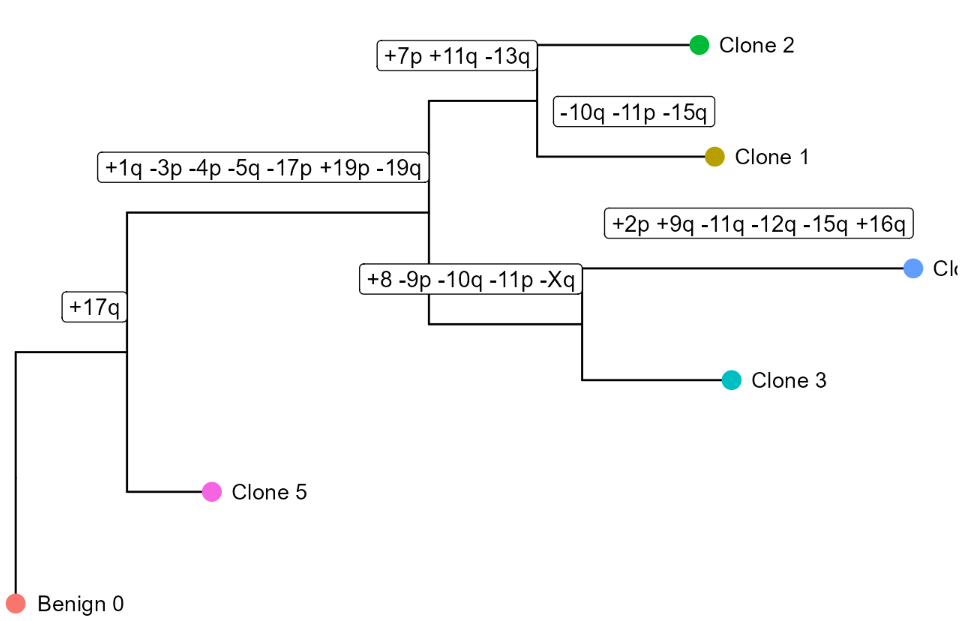
速度确实快啊,不过得测试测试
准确性似乎也更高?