Process Variability Source Analysis for a Multi-step Bio-process
Mario R. Eden, Marianthi Ierapetritou and Gavin P. Towler (Editors) Proceedings of the 13 th
International Symposium on Process Systems Engineering -- PSE 2018
July 1-5, 2018, San Diego, California, USA © 2018 Elsevier B.V. All rights reserved.
https://doi.org/10.1016/B978-0-444-64241-7.50411-0
目录
[2.1 高乳酸问题不是"一种"原因,至少存在4类不同机制](#2.1 高乳酸问题不是“一种”原因,至少存在4类不同机制)
[2.2 第2类高乳酸的主要根因指向接种阶段,且与两项可调控参数强相关](#2.2 第2类高乳酸的主要根因指向接种阶段,且与两项可调控参数强相关)
[2.3 可以在生产前预测第2类高乳酸风险,实现早期干预](#2.3 可以在生产前预测第2类高乳酸风险,实现早期干预)
[2.4 不同高乳酸聚类在细胞体积(PCV) 模式上存在显著差异](#2.4 不同高乳酸聚类在细胞体积(PCV) 模式上存在显著差异)
[2.5 未来可拓展的工艺数据采集建议(来自文章展望)](#2.5 未来可拓展的工艺数据采集建议(来自文章展望))
[2.6 总结:业务侧可执行的3条核心建议](#2.6 总结:业务侧可执行的3条核心建议)
[1. 适用范围](#1. 适用范围)
[2. 输入数据要求](#2. 输入数据要求)
[3. 软件与工具](#3. 软件与工具)
[4. 分析步骤](#4. 分析步骤)
[5. 注意事项](#5. 注意事项)
[7. 附录:历史研究贡献与局限](#7. 附录:历史研究贡献与局限)
一、问题背景与目标
-
背景 :在CHO细胞生产单克隆抗体的多步批次过程中,高乳酸 常与低产量相关,是影响工艺稳定性和产品质量的关键因素。
-
挑战:传统基于代谢机制的方法难以应用于大规模生产,因为缺少关键代谢物浓度数据。
-
目标 :利用历史数据挖掘高乳酸的根本原因 ,并实现在早期阶段(接种阶段) 的预测与干预。
二、业务侧结论
2.1 高乳酸问题不是"一种"原因,至少存在4类不同机制
结论 :
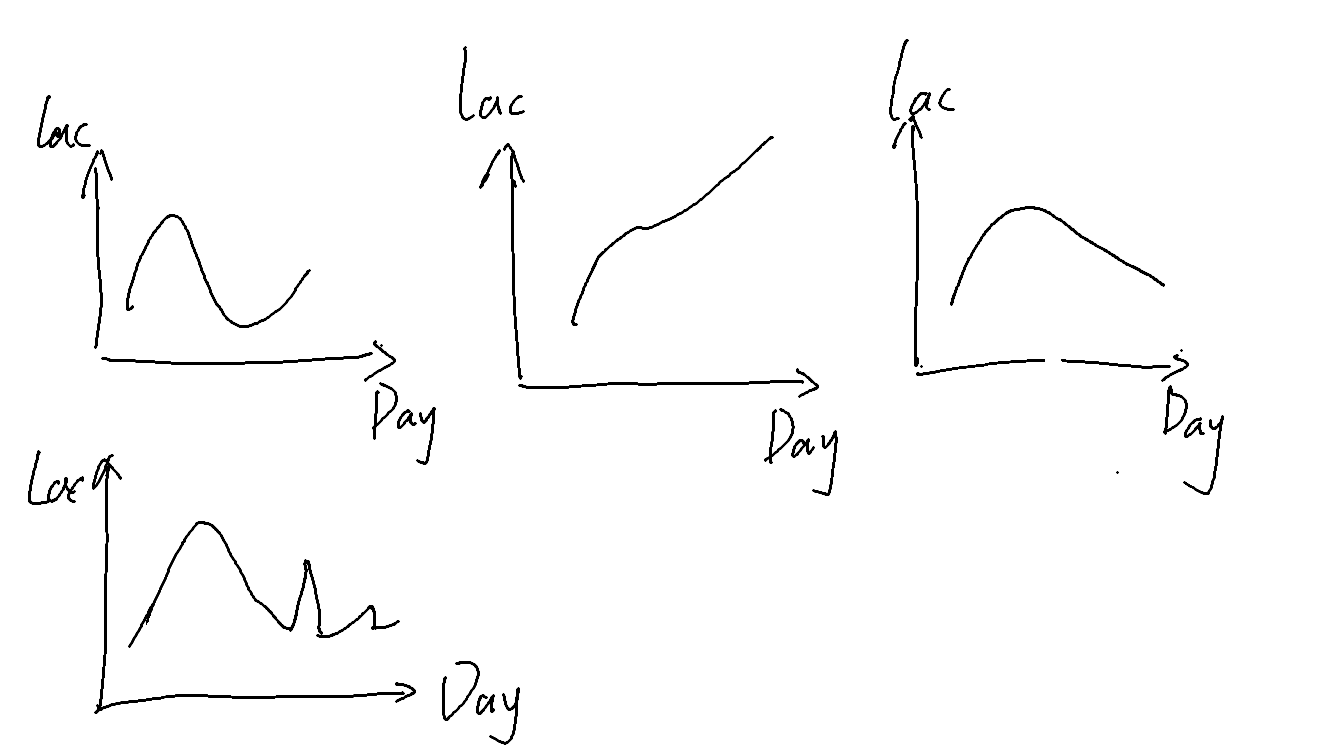
通过对241批生产数据的无监督聚类分析,高乳酸(低性能)批次可以被划分为 4个不同的子类,每个子类在关键生理参数(如PCV、代谢物)上表现出不同的演变模式。
按照我以往项目经验,有以下几种(第二行第一个是通过控糖来抑制lac翘尾)。注:与文中定义第几类的顺序不一样。
-
不要试图用一个"通用低乳酸方案"解决所有高乳酸批次。
-
建议针对每一类高乳酸设计独立的根因排查实验(例如:某类重点看接种条件,其他类可能涉及其他阶段或原料差异)。
2.2 第2类高乳酸的主要根因指向接种阶段,且与两项可调控参数强相关
结论:
-
第2类高乳酸(约占部分高乳酸批次)可以在接种(N-1 train)阶段被提前识别。
-
关键驱动因素是:
-
✅ 低 Na₂CO₃ / 培养基体积(即碳酸钠添加量偏低)
-
✅ 低初始渗透压
-
建议:
-
在接种阶段严格监控并优化这两个参数:
-
适当提高Na₂CO₃添加量(注意控制pH冲击)
-
提高初始渗透压至合适范围(可通过补加NaCl或调整培养基配方)
-
-
建议开展DOE实验,在N-1阶段验证这两个参数的调整对生产阶段乳酸积累的影响。
2.3 可以在生产前预测第2类高乳酸风险,实现早期干预
结论 :
使用接种阶段的23个可调节特征 + LDA降维 + SVM分类器,可以提前预测第2类高乳酸,模型性能:
-
误报率(Type I error)仅 2.3%
-
漏报率(Type II error)为 20.6%
建议:
-
可将该模型部署为工艺早期预警工具:在接种阶段结束后即可输出"是否属于第2类高乳酸高风险批次"。
-
对于高风险批次,可提前采取干预措施(如调整补料策略、温度控制、培养基优化)。
-
漏报率仍较高,建议结合其他指标(如PCV变化趋势)做综合判断,而非完全依赖模型。
2.4 不同高乳酸聚类在细胞体积(PCV) 模式上存在显著差异
结论 :
热图分析显示,第2类高乳酸的PCV演变模式与其他类明显不同(例如早期PCV偏低或上升过慢)。
建议:
-
将PCV曲线形态作为辅助诊断指标。
-
对于PCV模式类似第2类的批次,提前关注接种阶段渗透压和Na₂CO₃。
-
对于其他PCV模式的高乳酸批次,需另外设计实验(如代谢物分析、培养基批次差异)。
2.5 未来可拓展的工艺数据采集建议(来自文章展望)
结论 :
目前仅分析了接种阶段和生产阶段数据,但高乳酸的其他聚类可能关联到更早期的细胞解冻数据 或原材料(培养基/补料)批次信息。
建议:
-
建议工艺团队系统记录并共享以下数据给算法团队:
-
细胞解冻日期、活率、密度
-
培养基/补料的批次号、生产厂家、储存时间
-
接种前的细胞传代历史
-
-
这些数据可能解释目前尚无法归因的第1、3、4类高乳酸原因。
2.6 总结:业务侧可执行的3条核心建议
| 序号 | 核心建议 | 对应聚类 | 可行措施 |
|---|---|---|---|
| 1 | 接种阶段提高Na₂CO₃添加量 和初始渗透压 | 第2类 | 调整培养基配方或补料策略 |
| 2 | 部署早期SVM预警模型,识别第2类高风险批次 | 第2类 | 接种结束后运行模型,提前干预 |
| 3 | 对不同高乳酸类型分别设计根因实验,不要统一处理 | 全部4类 | 分类汇总历史批次,设计针对性DOE |
Lac根因分析流程
1. 适用范围
本文适用于CHO细胞培养生产单克隆抗体的多步骤批次工艺,基于历史生产数据(含生产阶段在线/离线测量、接种阶段数据、培养基制备数据)进行:
-
高乳酸与低乳酸批次的区分
-
高乳酸批次的隐藏模式识别聚类
-
接种阶段早期预测模型建立
-
潜在可调控根因识别
2. 输入数据要求
| 数据类型 | 说明 |
|---|---|
| 生产阶段数据(intra-batch) | 每个批次多个特征(如PCV、乳酸、葡萄糖等),每天采样2次,共23个时间点 |
| 接种阶段数据(N-1 train) | 每个批次初始状态、离线测量、培养基制备相关特征(至少23个选定特征) |
| 批次标签 | 最终乳酸浓度实测值,用于归一化及分类 |
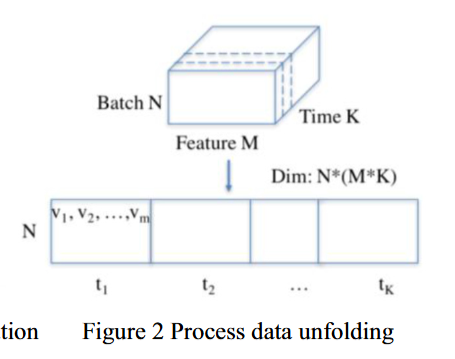
数据范围:至少包含2008-2015年间的历史批次,建议不少于200批。
数据模板参考如下:
3. 软件与工具
-
Python 3.8+ 或 MATLAB R2018b+
-
必需库:numpy, pandas, scikit-learn, matplotlib, seaborn
4. 分析步骤
python
import numpy as np
import pandas as pd
from sklearn.preprocessing import StandardScaler
from sklearn.decomposition import PCA
from sklearn.cluster import AgglomerativeClustering
from sklearn.discriminant_analysis import LinearDiscriminantAnalysis
from sklearn.svm import SVC
from scipy.cluster.hierarchy import linkage, dendrogram, fcluster
from scipy.stats import f, chi2
import matplotlib.pyplot as plt
from typing import Tuple, Optional, Dict, Any步骤1:数据预处理
-
缺失值处理 :对每个特征的时间序列,采用局部线性插值填补缺失点。
-
时间点对齐:确保每个批次具有相同的采样时间点(23个点)。
-
截取关键时间窗口:仅保留前13个时间点(高乳酸不可逆累积发生前的阶段)。
-
乳酸归一化:
-
计算所有批次最终乳酸浓度的最小值 Lmin和最大值 Lmax。
-
归一化值 Lnorm=(L−Lmin)/(Lmax−Lmin)。
-
分类阈值:Lnorm>0.4 为高乳酸批次(低性能),否则为低乳酸批次(高性能)。
-
python
def preprocess_and_normalize_lactate(
lactate_final: np.ndarray,
threshold: float = 0.4
) -> Tuple[np.ndarray, np.ndarray]:
"""
对最终乳酸浓度进行归一化,并根据阈值划分高/低乳酸批次。
Parameters
----------
lactate_final : np.ndarray, shape (n_batches,)
每个批次的最终乳酸浓度实测值。
threshold : float, default=0.4
归一化后的乳酸阈值,大于该值为高乳酸(低性能),否则为低乳酸(高性能)。
Returns
-------
lactate_norm : np.ndarray, shape (n_batches,)
归一化后的乳酸浓度,范围 [0, 1]。
high_lactate_mask : np.ndarray, shape (n_batches,), dtype=bool
True 表示高乳酸批次,False 表示低乳酸批次。
"""
L_min = np.min(lactate_final)
L_max = np.max(lactate_final)
lactate_norm = (lactate_final - L_min) / (L_max - L_min)
high_lactate_mask = lactate_norm > threshold
return lactate_norm, high_lactate_mask步骤2:数据展开(生产阶段数据)
-
原始数据形状:N 批次 × M 特征 × K 时间点(K=13)
-
展开为2D矩阵:每行对应一个批次,每列对应"某一特征在某一时间点"的值,总列数为 M×K。
-
示例:若 M=10 个特征,K=13,则展开后为 N×130 矩阵。
python
def unfold_3d_to_2d(
data_3d: np.ndarray,
) -> np.ndarray:
"""
将三维批次数据 (N, M, K) 展开为二维矩阵 (N, M*K)。
Parameters
----------
data_3d : np.ndarray, shape (n_batches, n_features, n_timepoints)
三维原始数据:批次 × 特征 × 时间点。
Returns
-------
data_2d : np.ndarray, shape (n_batches, n_features * n_timepoints)
展开后的二维矩阵,每列对应一个特征在某个时间点的值。
"""
n_batches, n_features, n_timepoints = data_3d.shape
return data_3d.reshape(n_batches, n_features * n_timepoints)步骤3:PCA监控模型(基于低乳酸批次)
-
训练数据:所有低乳酸批次(166批)的展开矩阵。
-
PCA拟合:
-
数据中心化(减去均值),是否缩放取决于特征单位,建议自动缩放(标准化)。
-
选择主成分数(PCs),使得累计方差解释率 ≥ 95%。【注:按照我的项目经验,基本上前3个主成分的累计方差解释率只有50%左右】
-
-
计算控制限:
-
T2 控制限:基于F分布(Qin 2003方法)。
-
SPE(Q统计量)控制限:基于卡方分布加权。
-
-
对所有批次(高+低乳酸)计算:
-
在PCA模型下投影,得到每个批次的 T2 和 SPE 值。
-
绘制散点图:T2 vs SPE。
-
观察:高乳酸与低乳酸批次在SPE(残差空间) 上分离明显。
-
python
def fit_pca_monitoring(
train_data: np.ndarray,
variance_threshold: float = 0.95,
alpha: float = 0.01
) -> Dict[str, Any]:
"""
在低乳酸批次上拟合PCA监控模型,并计算T2和SPE的控制限。
Parameters
----------
train_data : np.ndarray, shape (n_low_lactate_batches, n_features)
低乳酸批次的展开数据(已中心化/标准化,建议使用StandardScaler)。
variance_threshold : float, default=0.95
主成分累计方差解释率阈值,用于选择PC个数。
alpha : float, default=0.01
控制限的显著性水平(1 - 置信度)。
Returns
-------
model : dict
包含以下键的字典:
- 'pca': 拟合好的PCA对象
- 'scaler': 用于训练数据的StandardScaler对象(若输入已标准化可为None)
- 'T2_limit': T2统计量的控制限
- 'SPE_limit': SPE统计量的控制限
- 'n_components': 选定的主成分个数
"""
scaler = StandardScaler()
X_scaled = scaler.fit_transform(train_data)
pca = PCA(n_components=variance_threshold)
pca.fit(X_scaled)
n_comp = pca.n_components_
scores = pca.transform(X_scaled)
# 计算T2控制限 (基于F分布)
n = train_data.shape[0]
T2_limit = (n_comp * (n**2 - 1) / (n * (n - n_comp))) * f.ppf(1 - alpha, n_comp, n - n_comp)
# 计算SPE控制限 (基于卡方分布的加权近似,使用Qin 2003方法)
residuals = X_scaled - pca.inverse_transform(scores)
spe_vals = np.sum(residuals**2, axis=1)
mean_spe = np.mean(spe_vals)
var_spe = np.var(spe_vals)
g = var_spe / (2 * mean_spe)
h = 2 * (mean_spe**2) / var_spe
SPE_limit = g * chi2.ppf(1 - alpha, h)
return {
'pca': pca,
'scaler': scaler,
'T2_limit': T2_limit,
'SPE_limit': SPE_limit,
'n_components': n_comp
}
def calculate_pca_indices(
data: np.ndarray,
pca_model: Dict[str, Any]
) -> Tuple[np.ndarray, np.ndarray]:
"""
对新的批次数据计算T2和SPE统计量。
Parameters
----------
data : np.ndarray, shape (n_batches, n_features)
待计算的批次数据(需与训练数据特征数一致)。
pca_model : dict
由 fit_pca_monitoring 返回的模型字典。
Returns
-------
T2 : np.ndarray, shape (n_batches,)
每个批次的T2统计量。
SPE : np.ndarray, shape (n_batches,)
每个批次的SPE统计量(平方预测误差)。
"""
scaler = pca_model['scaler']
pca = pca_model['pca']
X_scaled = scaler.transform(data)
scores = pca.transform(X_scaled)
residuals = X_scaled - pca.inverse_transform(scores)
# T2
Lambda_inv = np.diag(1.0 / pca.explained_variance_)
T2 = np.sum(scores @ Lambda_inv * scores, axis=1)
# SPE
SPE = np.sum(residuals**2, axis=1)
return T2, SPE步骤4:高乳酸批次的层次聚类
-
提取数据:仅高乳酸批次(75批)在PCA残差空间中的得分(即原始数据减去PCA重构后的残差向量)。
-
层次聚类:
-
使用欧氏距离,Ward链接法。
-
绘制树状图。
-
使用肘部法(Within-Cluster Sum of Squares, WSS)确定最佳聚类数 k。
-
计算 k=1 到 10 的WSS。
-
选择WSS下降拐点对应的 k。论文结果为 k=4。【注:实际上4附近的线段弧度较为平滑,并非拐点。】
-
-
python
def hierarchical_clustering_residuals(
residuals: np.ndarray,
max_clusters: int = 10,
method: str = 'ward'
) -> Dict[str, Any]:
"""
对高乳酸批次的残差空间数据进行层次聚类,并自动确定最佳聚类数(肘部法)。
Parameters
----------
residuals : np.ndarray, shape (n_high_lactate, n_residual_features)
高乳酸批次在PCA残差空间中的值(通常使用残差得分或直接残差向量)。
max_clusters : int, default=10
肘部法考虑的最大聚类数。
method : str, default='ward'
层次聚类使用的链接方法(scipy.cluster.hierarchy.linkage 支持的方法)。
Returns
-------
result : dict
包含以下键的字典:
- 'Z': 链接矩阵(用于树状图)
- 'optimal_k': 肘部法确定的最佳聚类数
- 'cluster_labels': 每个批次的聚类标签 (1..optimal_k)
- 'wss': 每个k对应的组内平方和列表
"""
Z = linkage(residuals, method=method)
# 计算肘部法曲线
wss = []
for k in range(1, max_clusters + 1):
labels = fcluster(Z, t=k, criterion='maxclust')
cluster_centers = np.array([residuals[labels == i].mean(axis=0) for i in range(1, k+1)])
total_wss = sum(np.sum((residuals[labels == i] - cluster_centers[i-1])**2) for i in range(1, k+1))
wss.append(total_wss)
# 寻找拐点:一阶差分最小化(简化的肘部法,实际可结合斜率变化)
diffs = np.diff(wss)
optimal_k = np.argmin(diffs) + 1 # 最简单的拐点选择
# 使用最优k进行聚类
cluster_labels = fcluster(Z, t=optimal_k, criterion='maxclust')
return {
'Z': Z,
'optimal_k': optimal_k,
'cluster_labels': cluster_labels,
'wss': wss
}聚类可视化:
-
使用LDA将聚类结果降维到2D或3D空间,绘制散点图,观察各聚类分离情况。
-
绘制热图(heatmap)展示每个聚类在关键特征(如PCV)上的残差模式。
python
def reduce_dimension_lda(
features: np.ndarray,
labels: np.ndarray,
n_components: int = 3
) -> Dict[str, Any]:
"""
使用线性判别分析 (LDA) 对特征降维,用于可视化或后续分类。
Parameters
----------
features : np.ndarray, shape (n_samples, n_features)
原始特征矩阵(如接种阶段的23个可调控特征)。
labels : np.ndarray, shape (n_samples,)
类别标签(0/1 或更多多类标签)。
n_components : int, default=3
降维后的维度,必须小于类别数。
Returns
-------
model : dict
包含以下键的字典:
- 'lda': 拟合好的LDA对象
- 'X_lda': 降维后的数据,shape (n_samples, n_components)
- 'explained_variance_ratio': 每个LD的解释方差比例(若可用)
"""
lda = LinearDiscriminantAnalysis(n_components=n_components)
X_lda = lda.fit_transform(features, labels)
return {
'lda': lda,
'X_lda': X_lda,
'explained_variance_ratio': lda.explained_variance_ratio_ if hasattr(lda, 'explained_variance_ratio_') else None
}步骤5:接种阶段特征筛选与降维
-
特征选择 :基于工艺知识,从接种阶段数据中筛选出可调控的23个特征(如初始渗透压、Na₂CO₃/培养基体积比、接种密度等)。
-
标签定义:
- 将步骤4中聚类得到的某个特定子类(如cluster 2)标记为"异常"(1),其余所有批次(包括低乳酸及其他高乳酸子类)标记为"正常"(0)。
-
LDA降维:
-
对23个特征进行LDA,目标维度为3(得到LD1, LD2, LD3)。
-
可视化LD1-LD2和LD1-LD3散点图,观察"异常"与"正常"的可分性。
-
步骤6:SVM分类模型训练与评估
-
输入:LDA降维后的前3个线性判别(LDs)。
-
模型:SVM,核函数为RBF,参数 γ=2γ=2(论文设定)。可使用网格搜索微调。
-
训练:全部数据(因样本量不大,未明确划分训练/测试集,但可做交叉验证)。
-
性能评估:
-
计算混淆矩阵。
-
Type I error(假阳性率):正常批次被误判为异常的比例。
-
Type II error(假阴性率):异常批次被漏判的比例。
-
论文结果:Type I error = 2.3%,Type II error = 20.6%。
-
python
def train_svm_classifier(
X_train: np.ndarray,
y_train: np.ndarray,
kernel: str = 'rbf',
gamma: float = 2.0,
**kwargs
) -> SVC:
"""
训练SVM分类器用于早期异常批次识别。
Parameters
----------
X_train : np.ndarray, shape (n_samples, n_features)
训练数据特征(通常为LDA降维后的前3个LD)。
y_train : np.ndarray, shape (n_samples,)
训练标签(1为异常,0为正常)。
kernel : str, default='rbf'
SVM核函数类型。
gamma : float, default=2.0
RBF核的参数γ。
**kwargs : dict
传递给 sklearn.svm.SVC 的其他参数。
Returns
-------
svm_model : sklearn.svm.SVC
训练好的SVM分类器。
"""
svm = SVC(kernel=kernel, gamma=gamma, **kwargs)
svm.fit(X_train, y_train)
return svm
def evaluate_classifier(
model: SVC,
X_test: np.ndarray,
y_test: np.ndarray
) -> Dict[str, float]:
"""
评估分类器性能,计算Type I和Type II错误率。
Parameters
----------
model : sklearn.svm.SVC
训练好的分类器。
X_test : np.ndarray, shape (n_samples, n_features)
测试数据。
y_test : np.ndarray, shape (n_samples,)
真实标签。
Returns
-------
metrics : dict
包含以下键的字典:
- 'accuracy': 准确率
- 'type1_error': 假阳性率(正常被误判为异常)
- 'type2_error': 假阴性率(异常被漏判)
- 'confusion_matrix': 混淆矩阵 (2x2)
"""
from sklearn.metrics import confusion_matrix
y_pred = model.predict(X_test)
tn, fp, fn, tp = confusion_matrix(y_test, y_pred, labels=[0, 1]).ravel()
type1_error = fp / (fp + tn) if (fp + tn) > 0 else 0.0
type2_error = fn / (fn + tp) if (fn + tp) > 0 else 0.0
accuracy = (tp + tn) / (tp + tn + fp + fn)
return {
'accuracy': accuracy,
'type1_error': type1_error,
'type2_error': type2_error,
'confusion_matrix': [[tn, fp], [fn, tp]]
}步骤7:根因识别与可视化
-
针对区分出的特定子类(如cluster 2),比较其与正常批次在原始接种阶段特征上的分布差异。
-
绘制箱线图(box plot)展示关键特征(如Na₂CO₃/体积、初始渗透压)在两组的对比。
-
识别出差异显著且工艺可调的特征作为潜在根因。
python
def plot_boxplot_comparison(
data: pd.DataFrame,
feature_names: list,
group_labels: np.ndarray,
group_names: Tuple[str, str] = ('Normal', 'Abnormal'),
save_path: Optional[str] = None
):
"""
绘制箱线图对比正常组与异常组在特定特征上的分布。
Parameters
----------
data : pd.DataFrame, shape (n_samples, n_features)
包含所有样本和特征的DataFrame。
feature_names : list
需要绘制的特征名称列表。
group_labels : np.ndarray, shape (n_samples,)
每组标签(0:正常, 1:异常)。
group_names : tuple, default=('Normal', 'Abnormal')
图例中的组名称。
save_path : str, optional
如果提供,则保存图像到该路径。
"""
n_features = len(feature_names)
fig, axes = plt.subplots(1, n_features, figsize=(5 * n_features, 4))
if n_features == 1:
axes = [axes]
for ax, feat in zip(axes, feature_names):
data_to_plot = [data.loc[group_labels == 0, feat].dropna(),
data.loc[group_labels == 1, feat].dropna()]
bp = ax.boxplot(data_to_plot, labels=group_names, patch_artist=True)
ax.set_title(feat)
ax.set_ylabel('Value')
ax.grid(True, linestyle='--', alpha=0.7)
plt.tight_layout()
if save_path:
plt.savefig(save_path, dpi=300)
plt.show()5. 注意事项
-
数据必须来自相同工艺平台(培养基、接种流程、培养条件一致),否则需重新建模。
-
缺失值处理应谨慎,若某特征缺失率>20%,建议剔除。
-
聚类数 k 的确定需结合工艺可解释性,不唯WSS。
-
早期预测模型(SVM)仅适用于被识别出的特定异常子类,其他子类需单独建模。
-
本文未包含时间序列的动态轨迹监控(如multi-way PCA),如需在线监控,需扩展方法。
6. DEMO.py
python
# test_demo.py
"""
简易测试脚本:生成模拟数据,验证多步骤生物工艺变异性分析模块
使用方法:python test_demo.py
要求:已安装 numpy, pandas, scikit-learn, scipy, matplotlib
"""
import numpy as np
import pandas as pd
from sklearn.preprocessing import StandardScaler
from sklearn.decomposition import PCA
from scipy.cluster.hierarchy import linkage, fcluster
from sklearn.discriminant_analysis import LinearDiscriminantAnalysis
from sklearn.svm import SVC
from sklearn.metrics import confusion_matrix
import matplotlib
matplotlib.use('Agg') # 无图形界面时使用,保存图片
import matplotlib.pyplot as plt
from typing import Tuple, Dict, Any
# ---------- 1. 导入之前定义的函数(假设它们已保存在 module.py 中,这里直接复制定义以保证独立运行) ----------
# 实际使用时可 from your_module import *
# 为了demo独立,将核心函数拷贝如下(可省略部分注释以节省篇幅)
def preprocess_and_normalize_lactate(lactate_final: np.ndarray, threshold: float = 0.4
) -> Tuple[np.ndarray, np.ndarray]:
L_min, L_max = np.min(lactate_final), np.max(lactate_final)
lactate_norm = (lactate_final - L_min) / (L_max - L_min)
high_lactate_mask = lactate_norm > threshold
return lactate_norm, high_lactate_mask
def unfold_3d_to_2d(data_3d: np.ndarray) -> np.ndarray:
n_batches, n_features, n_timepoints = data_3d.shape
return data_3d.reshape(n_batches, n_features * n_timepoints)
def fit_pca_monitoring(train_data: np.ndarray, variance_threshold: float = 0.95, alpha: float = 0.01
) -> Dict[str, Any]:
scaler = StandardScaler()
X_scaled = scaler.fit_transform(train_data)
pca = PCA(n_components=variance_threshold)
pca.fit(X_scaled)
n_comp = pca.n_components_
scores = pca.transform(X_scaled)
n = train_data.shape[0]
from scipy.stats import f, chi2
T2_limit = (n_comp * (n**2 - 1) / (n * (n - n_comp))) * f.ppf(1 - alpha, n_comp, n - n_comp)
residuals = X_scaled - pca.inverse_transform(scores)
spe_vals = np.sum(residuals**2, axis=1)
mean_spe, var_spe = np.mean(spe_vals), np.var(spe_vals)
g = var_spe / (2 * mean_spe)
h = 2 * (mean_spe**2) / var_spe
SPE_limit = g * chi2.ppf(1 - alpha, h)
return {'pca': pca, 'scaler': scaler, 'T2_limit': T2_limit, 'SPE_limit': SPE_limit, 'n_components': n_comp}
def calculate_pca_indices(data: np.ndarray, pca_model: Dict[str, Any]) -> Tuple[np.ndarray, np.ndarray]:
scaler = pca_model['scaler']
pca = pca_model['pca']
X_scaled = scaler.transform(data)
scores = pca.transform(X_scaled)
residuals = X_scaled - pca.inverse_transform(scores)
Lambda_inv = np.diag(1.0 / pca.explained_variance_)
T2 = np.sum((scores @ Lambda_inv) * scores, axis=1)
SPE = np.sum(residuals**2, axis=1)
return T2, SPE
def hierarchical_clustering_residuals(residuals: np.ndarray, max_clusters: int = 10, method: str = 'ward'
) -> Dict[str, Any]:
Z = linkage(residuals, method=method)
wss = []
for k in range(1, max_clusters + 1):
labels = fcluster(Z, t=k, criterion='maxclust')
centers = np.array([residuals[labels == i].mean(axis=0) for i in range(1, k+1)])
total_wss = sum(np.sum((residuals[labels == i] - centers[i-1])**2) for i in range(1, k+1))
wss.append(total_wss)
diffs = np.diff(wss)
optimal_k = np.argmin(diffs) + 1
cluster_labels = fcluster(Z, t=optimal_k, criterion='maxclust')
return {'Z': Z, 'optimal_k': optimal_k, 'cluster_labels': cluster_labels, 'wss': wss}
def reduce_dimension_lda(features: np.ndarray, labels: np.ndarray, n_components: int = 3) -> Dict[str, Any]:
lda = LinearDiscriminantAnalysis(n_components=n_components)
X_lda = lda.fit_transform(features, labels)
return {'lda': lda, 'X_lda': X_lda, 'explained_variance_ratio': getattr(lda, 'explained_variance_ratio_', None)}
def train_svm_classifier(X_train: np.ndarray, y_train: np.ndarray, kernel: str = 'rbf', gamma: float = 2.0, **kwargs):
svm = SVC(kernel=kernel, gamma=gamma, **kwargs)
svm.fit(X_train, y_train)
return svm
def evaluate_classifier(model, X_test: np.ndarray, y_test: np.ndarray) -> Dict[str, float]:
y_pred = model.predict(X_test)
tn, fp, fn, tp = confusion_matrix(y_test, y_pred, labels=[0, 1]).ravel()
type1_error = fp / (fp + tn) if (fp + tn) > 0 else 0.0
type2_error = fn / (fn + tp) if (fn + tp) > 0 else 0.0
accuracy = (tp + tn) / (tp + tn + fp + fn)
return {'accuracy': accuracy, 'type1_error': type1_error, 'type2_error': type2_error,
'confusion_matrix': [[tn, fp], [fn, tp]]}
def plot_boxplot_comparison(data: pd.DataFrame, feature_names: list, group_labels: np.ndarray,
group_names: Tuple[str, str] = ('Normal', 'Abnormal'), save_path: str = None):
n_feat = len(feature_names)
fig, axes = plt.subplots(1, n_feat, figsize=(5 * n_feat, 4))
if n_feat == 1:
axes = [axes]
for ax, feat in zip(axes, feature_names):
data_to_plot = [data.loc[group_labels == 0, feat].dropna(), data.loc[group_labels == 1, feat].dropna()]
ax.boxplot(data_to_plot, labels=group_names, patch_artist=True)
ax.set_title(feat)
ax.set_ylabel('Value')
ax.grid(True, linestyle='--', alpha=0.7)
plt.tight_layout()
if save_path:
plt.savefig(save_path, dpi=300)
print(f"Boxplot saved to {save_path}")
plt.close(fig)
# ---------- 2. 生成模拟数据 ----------
print("Step 0: Generating synthetic data...")
np.random.seed(42)
n_batches = 241
n_features_intra = 10 # 生产阶段特征数,如 PCV, 乳酸, 葡萄糖等
n_timepoints = 13 # 前13个时间点
n_features_inoc = 23 # 接种阶段可调控特征数
# 生产阶段数据:模拟正常批次 + 部分异常批次
X_intra = np.random.randn(n_batches, n_features_intra, n_timepoints) * 0.5
# 为高乳酸批次添加偏移(模拟异常)
high_idx = np.arange(75) # 假设前75个为高乳酸,后面正常
X_intra[high_idx, :, 5:] += 0.8 # 后期时间点偏移
# 接种阶段数据:正常批次均值0,高乳酸批次在部分特征上有偏移
X_inoc = np.random.randn(n_batches, n_features_inoc) * 0.3
# 让其中一部分高乳酸批次(例如聚类2)在特征0和特征1上有显著偏移
X_inoc[high_idx[:30], 0] -= 1.2 # 低 Na2CO3/体积
X_inoc[high_idx[:30], 1] -= 1.0 # 低初始渗透压
# 最终乳酸浓度:正常批次低,高乳酸批次高
lactate_final = np.random.randn(n_batches) * 0.2 + 0.5
lactate_final[high_idx] += 0.6
lactate_final = np.clip(lactate_final, 0.1, 1.2)
# ---------- 3. 运行分析流程 ----------
print("Step 1: Lactate normalization and split...")
lactate_norm, high_mask = preprocess_and_normalize_lactate(lactate_final, threshold=0.4)
n_high = np.sum(high_mask)
n_low = n_batches - n_high
print(f" Low lactate batches: {n_low}, High lactate batches: {n_high}")
print("Step 2: Unfold 3D intra-batch data...")
X_2d = unfold_3d_to_2d(X_intra) # shape (241, 10*13=130)
print("Step 3: Fit PCA on low-lactate batches...")
low_data = X_2d[~high_mask]
pca_model = fit_pca_monitoring(low_data, variance_threshold=0.95, alpha=0.01)
print(f" Number of PCs: {pca_model['n_components']}")
print(f" T2 limit: {pca_model['T2_limit']:.3f}, SPE limit: {pca_model['SPE_limit']:.3f}")
print("Step 4: Calculate T2 and SPE for all batches...")
T2_all, SPE_all = calculate_pca_indices(X_2d, pca_model)
print(f" T2 range: [{T2_all.min():.2f}, {T2_all.max():.2f}], SPE range: [{SPE_all.min():.2f}, {SPE_all.max():.2f}]")
print("Step 5: Extract residual space for high-lactate batches and perform clustering...")
scaler = pca_model['scaler']
pca = pca_model['pca']
high_data_scaled = scaler.transform(X_2d[high_mask])
high_scores = pca.transform(high_data_scaled)
high_residuals = high_data_scaled - pca.inverse_transform(high_scores) # shape (n_high, 130)
clustering_res = hierarchical_clustering_residuals(high_residuals, max_clusters=6)
optimal_k = clustering_res['optimal_k']
cluster_labels = clustering_res['cluster_labels']
print(f" Optimal number of clusters (elbow): {optimal_k}")
print(f" Cluster distribution: {np.bincount(cluster_labels)[1:]}")
print("Step 6: LDA on inoculum data using cluster 2 as abnormal...")
# 取高乳酸批次中属于 cluster 2 的作为异常,其余(低乳酸+其他高乳酸)作为正常
high_batch_indices = np.where(high_mask)[0]
abnormal_mask = np.zeros(n_batches, dtype=bool)
for idx, lbl in zip(high_batch_indices, cluster_labels):
if lbl == 2: # 假设 cluster 2 是我们关注的异常子类
abnormal_mask[idx] = True
y_labels = abnormal_mask.astype(int)
print(f" Abnormal batches (cluster 2): {np.sum(y_labels)}")
lda_res = reduce_dimension_lda(X_inoc, y_labels, n_components=3)
print(f" LDA explained variance ratio: {lda_res['explained_variance_ratio']}")
print("Step 7: Train SVM classifier on LDA-reduced features...")
X_lda = lda_res['X_lda']
svm = train_svm_classifier(X_lda, y_labels, kernel='rbf', gamma=2.0)
metrics = evaluate_classifier(svm, X_lda, y_labels) # 用相同数据评估(demo仅测试跑通)
print(f" SVM Accuracy: {metrics['accuracy']:.3f}, Type I error: {metrics['type1_error']:.3f}, Type II error: {metrics['type2_error']:.3f}")
print("Step 8: Generate boxplot for potential root causes...")
# 构建 DataFrame 并选取两个代表性特征
df_inoc = pd.DataFrame(X_inoc, columns=[f'Feat_{i}' for i in range(n_features_inoc)])
root_cause_features = ['Feat_0', 'Feat_1'] # 对应模拟的低Na2CO3和低渗透压
plot_boxplot_comparison(df_inoc, root_cause_features, y_labels, save_path='boxplot_demo.png')
print(" Boxplot saved as 'boxplot_demo.png'")
print("\nAll steps completed successfully! The module functions are working correctly.")7. 附录:历史研究贡献与局限
| 作者(年份) | 研究方法 / 模型 | 研究对象 / 目的 | 主要发现 / 贡献 | 局限性(文中指出) |
|---|---|---|---|---|
| Charaniya et al. (2010) | 数据挖掘(关联规则等) | CHO细胞培养过程,高产量工艺特征发现 | 发现低细胞生长与高乳酸、低产量之间的相关性 | 未在文中具体指出,但属于早期数据驱动尝试 |
| Mulukutla et al. (2015) | 稳态拓扑代谢模型(代谢网络分析) | 哺乳动物细胞糖酵解与乳酸代谢 | 解释乳酸积累与消耗的机制,指出代谢状态转移的可能性 | 需要额外的代谢物浓度数据,工业大规模生产中不常规检测 |
| Zagari et al. (2013) | 线粒体氧化活性分析 | CHO细胞培养中的乳酸代谢转变 | 揭示乳酸代谢转变与线粒体功能的关系 | 同样需要专门代谢分析,难以直接应用于商业生产过程监控 |
| Nomikos & MacGregor (1995) | 多向偏最小二乘法(mPLS) | 批次过程监测 | 提出mPLS用于提取轨迹数据信息,建立监测图表 | 方法本身为经典方法,但未直接针对高乳酸问题 |
| Nomikos (1996) | 多向主成分分析(mPCA) | 异常批次操作检测与诊断 | 基于mPCA的批次过程监控框架 | 主要用于故障检测,未深入分析多阶段早期原因 |
| Qin (2003, 2012) | 统计过程监控(PCA、SPE、T²等) | 工业过程监测与诊断 | 系统总结了PCA监控及控制限计算方法(如SPE、T²) | 属于基础方法学,未针对生物过程特定问题 |
说明 :上述表格仅涵盖本文引用的、与细胞培养过程变异性分析直接相关的历史研究。本文作者在现有研究基础上,提出将 PCA + 层次聚类 + SVM 结合,并利用接种阶段可调控特征进行早期预测,弥补了代谢模型难落地、传统MVA方法未深入多阶段根因分析的不足。