01
导语
各位同学,大家好。现在做影像组学,如果还只停留在"提取特征---建个模型---算个AUC",那就有点像算命算得挺准,但为啥准,自己也说不明白。别人一问:你这特征到底代表啥?背后有啥道理?瞬间就成了黑箱本箱 。而今天解读的这篇文献,虽然核心用的是转录组数据,但它展示了一套教科书级别的"机制挂靠"范式 :从随机森林模型LIRA出发,一路追到HDAC4去乙酰化通路 、LRP8-APOE免疫抑制信号轴 ,再用单细胞验证Treg和恶性细胞占比,最后用深度学习病理热图锁定淋巴细胞 vs. 肿瘤+成纤维细胞 的组织学差异。这告诉我们:真正能打高分的研究,都得给模型找"生物学娘家" 。咱们就来聊聊,影像组学如何借鉴这套思路,从"只会算分的工具人"升级成能讲清疾病故事的研究者。

**★题目:**Predicting Immunotherapy Outcomes in NSCLC Using RNA and Pathology from Multicenter Clinical Trials
(利用多中心临床试验的RNA和病理学数据预测非小细胞肺癌免疫治疗结局)
★期刊:《Advanced Science》(中科院1区,IF=14.1)
**★研究疾病:**晚期非小细胞肺癌
**★生物学机制:**去乙酰化促免疫逃逸
**★发表时间:**2025年10月
02
研究背景-从 "临床问题" 落到 "生物学问题"
晚期非小细胞肺癌(NSCLC)的治疗中,免疫检查点抑制剂(ICI)已成为无驱动基因突变患者的一线标准方案 。然而,临床实践显示,仅约15%--25%的患者能从ICI单药治疗中获得持久缓解,大部分患者面临原发性耐药或早期进展 的风险。目前临床最常用的预测生物标志物是肿瘤细胞PD-L1表达(TPS)和肿瘤突变负荷(TMB) ,但两者均存在明显局限:PD-L1表达具有时空异质性,且部分PD-L1阴性患者仍可获益;TMB作为抗原性的间接指标,在不同检测平台和样本类型间差异较大,且不能反映肿瘤微环境(TME)的免疫状态 。近年来,基于T细胞炎症基因表达谱(GEP)的生物标志物虽有所进展,但仍难以全面捕捉TME中免疫细胞、基质细胞、代谢通路及表观遗传调控之间的复杂交互作用。因此,如何从"单维度标志物"转向"整合性分子模型" ,以准确识别真正能从ICI治疗中获益的NSCLC患者,成为亟待解决的临床-生物学问题。本文正是立足于这一困境,试图通过转录组大数据与机器学习,构建一个能够反映TME免疫状态和耐药机制的预测工具。
03
研究目的(明确写出"三层目的")
本研究的设计具有清晰的三层目的 。第一层(临床层) :开发一个基于转录组数据的机器学习模型,命名为LIRA(肺癌免疫治疗反应评估) ,用于预测接受ICI治疗的晚期NSCLC患者的生存获益(无进展生存期和总生存期) ,并在多个独立的多中心随机对照试验(POPLAR、OAK、ORIENT‑11)及真实世界队列中进行严格验证。第二层(技术层) :通过随机生存森林算法 和严格的特征筛选流程(单因素Cox回归、治疗交互作用分析、变量重要性排序),从海量基因中选出50个与免疫治疗结局最相关的模型基因,并计算每位患者的LIRA评分;同时建立单样本预测参考数据库 ,使模型具备个体化临床应用能力。第三层(生物学层) :这也是本研究区别于大多数纯预测模型的核心创新------揭示LIRA评分背后的生物学机制 ,即解释"为什么高LIRA评分患者对ICI响应好,而低LIRA评分患者容易耐药"。通过差异表达分析、通路富集、配体-受体互作、单细胞转录组测序以及基于深度学习的病理全切片图像分析,将LIRA评分与免疫细胞浸润、乙酰化/去乙酰化修饰、LRP8-APOE信号轴、Treg比例及肿瘤-成纤维细胞互作等具体生物学过程挂钩,从而为模型的预测能力提供坚实的机制支撑。
04
研究思路(最核心:怎么挂靠机制)
本研究的整体思路体现了 " 从表型到机制"的多层次递进设计 ,其最值得借鉴之处在于将机器学习的预测评分系统性地锚定于生物学通路 。首先,利用OAK和POPLAR两大随机临床试验中891例患者的转录组数据,采用随机生存森林 构建LIRA模型,并在内部及外部验证队列中证明其预测PFS/OS的能力显著优于PD-L1 TPS和TMB。随后,机制挂靠 成为核心步骤:通过差异表达分析 发现低LIRA评分组显著富集组蛋白乙酰化/去乙酰化相关通路 ;进一步通过单变量Cox回归和Kaplan‑Meier分析 锁定HDAC4 作为关键不良预后基因;运用配体-受体分析 揭示低分组中LRP8-APOE 信号轴增强,提示免疫抑制性代谢微环境。为了从细胞层面验证,研究引入独立NSCLC单细胞RNA-seq数据 (41,799个细胞),发现低LIRA评分组中Treg细胞比例升高、恶性上皮细胞增多 ,而高分组则以T细胞、B细胞浸润为主。最后,通过基于CLAM框架的深度学习模型 分析TCGA的病理全切片图像,自动生成注意力热图,并由病理专家标注发现:高LIRA评分组的高关注区域主要为淋巴细胞 ,低分组则为肿瘤细胞和成纤维细胞 ,从组织学层面印证了转录组和单细胞层面的发现。这一思路将分子通路、细胞组成、组织形态三个层次的证据串联成完整的机制链条,使LIRA评分不再是一个"黑箱"分数,而是可解释的生物学指标。
05
数据和方法(机制部分怎么设计)
数据: 本研究整合了来自多中心随机临床试验 的1127例晚期NSCLC患者的转录组及临床数据。其中,OAK(n=699)和POPLAR(n=192)队列 共891例患者(接受atezolizumab单药或多西他赛化疗),按7:3随机划分为训练集(n=625)和内部验证集(n=266) ,用于LIRA模型的构建与初步验证。ORIENT-11队列(n=171) 和南方医院(NFH)真实世界队列(n=65) (接受PD-1抑制剂联合化疗或单药治疗)作为外部验证集 ,评估模型的泛化能力。此外,IMvigor210(尿路上皮癌)和IMpower133(小细胞肺癌) 队列用于跨癌种验证;TCGA-LUAD、TCGA-LUSC及多个GEO数据集 用于非免疫治疗场景分析。为探索机制,采用独立NSCLC单细胞RNA-seq数据集GSE131907(11例患者,41,799个细胞) 进行细胞层面验证,并利用**TCGA-LUAD/LUSC的病理全切片图像(WSI,共971例患者,1524张切片)**训练深度学习模型,以预测LIRA分组并生成注意力热图。
方法: ① RNA-seq 数据处理 :使用fastp质控、STAR比对至GRCh38,获得基因表达矩阵。② 特征基因筛选 :在训练集中依次进行单变量Cox回归 (p<0.01)、治疗交互作用分析 (p for interaction<0.05),再通过随机生存森林 选取变量重要性前50的基因 作为模型基因。③ LIRA 模型构建 :基于上述50个基因,采用随机生存森林算法计算每位患者的LIRA评分 ,以队列均值划分高/低评分组。④ 参考数据库建立 :汇总439例atezolizumab治疗患者的转录组数据,用于单样本预测 时去除批次效应并确定最佳截断值。⑤ 机制分析 :通过差异表达分析 、GO/GSEA通路富集 、ssGSEA计算乙酰化相关通路评分 ,识别关键基因HDAC4 和LRP8 ;利用EaSIeR 和CellChat 进行配体-受体分析。⑥ 单细胞分析 :采用Seurat降维聚类、InferCNV区分恶性/非恶性上皮细胞,比较高低LIRA组的细胞组成。⑦ 深度学习病理模型 :基于CLAM框架 ,提取WSI的10倍镜 patches(256×256),使用UNI基础模型 提取1024维特征,训练LIRA分类器 (三分类:高/中/低组),并生成注意力热图,由病理专家标注高贡献区域。
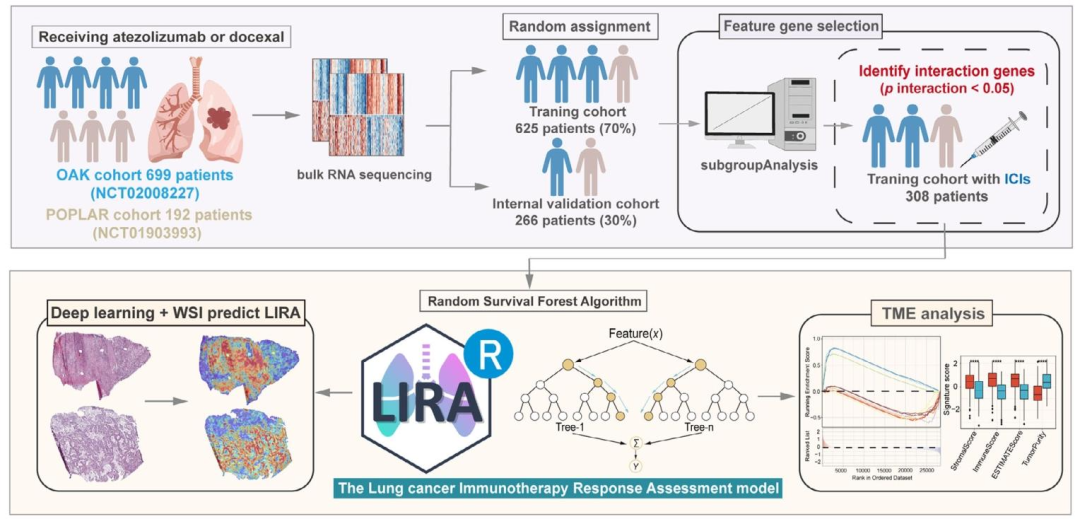
图 1:研究工作整体流程图
06
研究结果("从表型到机制")
1. LIRA在二/三线PD‑L1单抗治疗中精准预测生存获益
在POPLAR和OAK队列中,高LIRA评分组接受atezolizumab治疗的患者,PFS(HR=0.15)和OS(HR=0.34)显著延长;而低评分组从化疗中获益更优,证明LIRA可有效区分免疫治疗与化疗的优势人群。
2. LIRA在一线PD‑1联合化疗中同样有效
在ORIENT‑11和NFH队列中,高LIRA评分患者接受联合治疗后的PFS和OS均显著优于低评分患者;低评分组中免疫联合化疗对比单纯化疗无显著差异,提示低评分患者可能无需加用免疫治疗。
3. LIRA预测性能显著优于PD‑L1和TMB
时间依赖ROC分析显示,LIRA预测PFS的AUC(0.78-0.96)显著高于TPS(0.52-0.63)和tTMB(0.52-0.67);联合LIRA与TMB/TPS的模型未表现出统计学显著提升,表明LIRA单用已具高效费比。
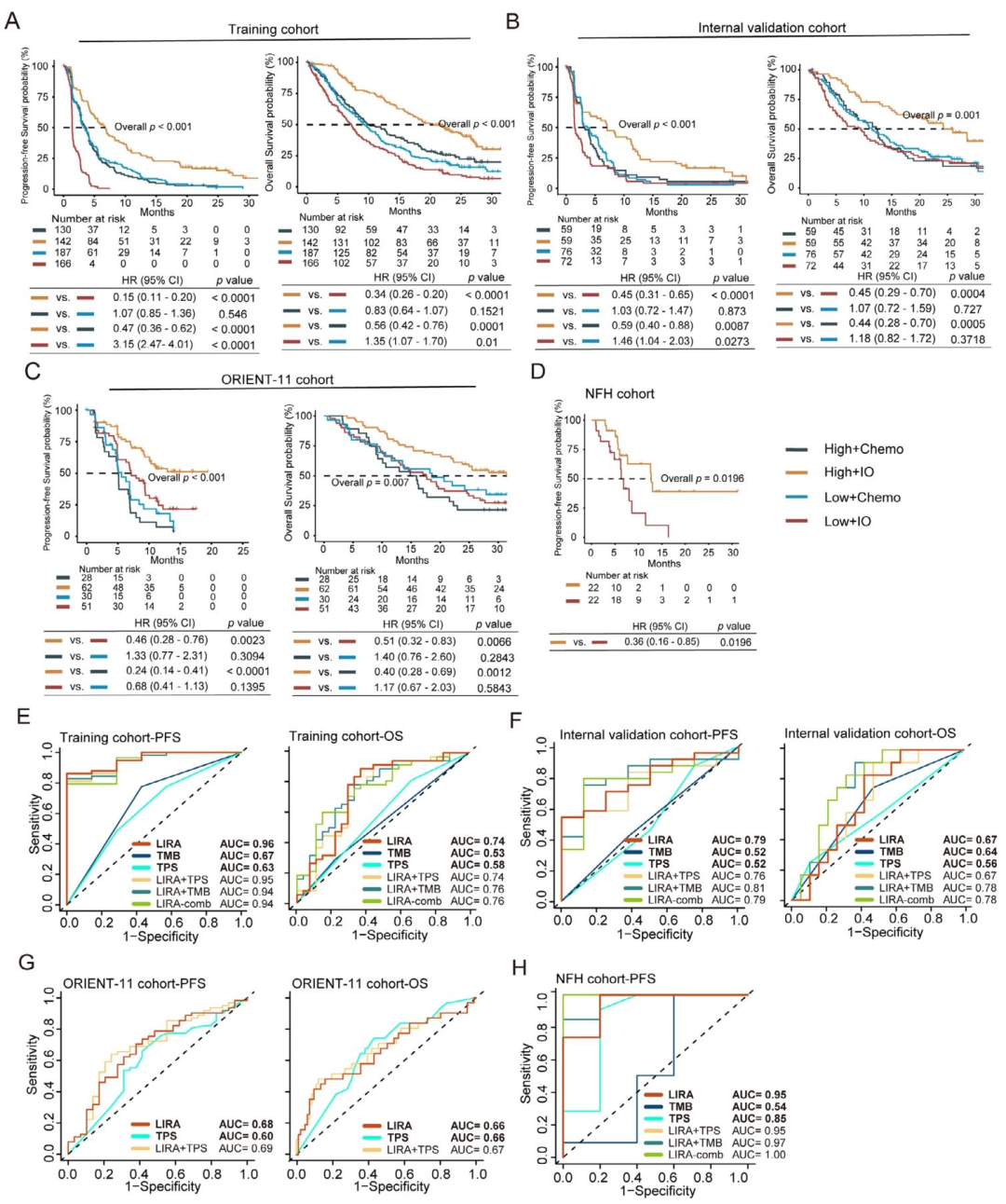
图 2(生存分析与ROC曲线) :图中A-D展示了高/低LIRA评分组在训练集、内部验证集、ORIENT‑11和NFH队列中的Kaplan‑Meier曲线,高评分组PFS和OS均显著优于低评分组 (HR低至0.15)。E-H为时间依赖ROC,LIRA的AUC(0.78-0.96)显著高于TPS和TMB。影像组学可借鉴:通过生存曲线证明影像标签的预后区分能力,再通过ROC对比证明其优于传统标志物。
4. LIRA评分独立于传统标志物且适用于驱动基因突变患者
多因素Cox回归证实,LIRA评分独立于TPS、tTMB、组织学类型和性别(HR=0.258)。在EGFR、STK11、KEAP1突变亚组中,LIRA仍能准确预测免疫治疗获益,说明其具有广泛的适用性。
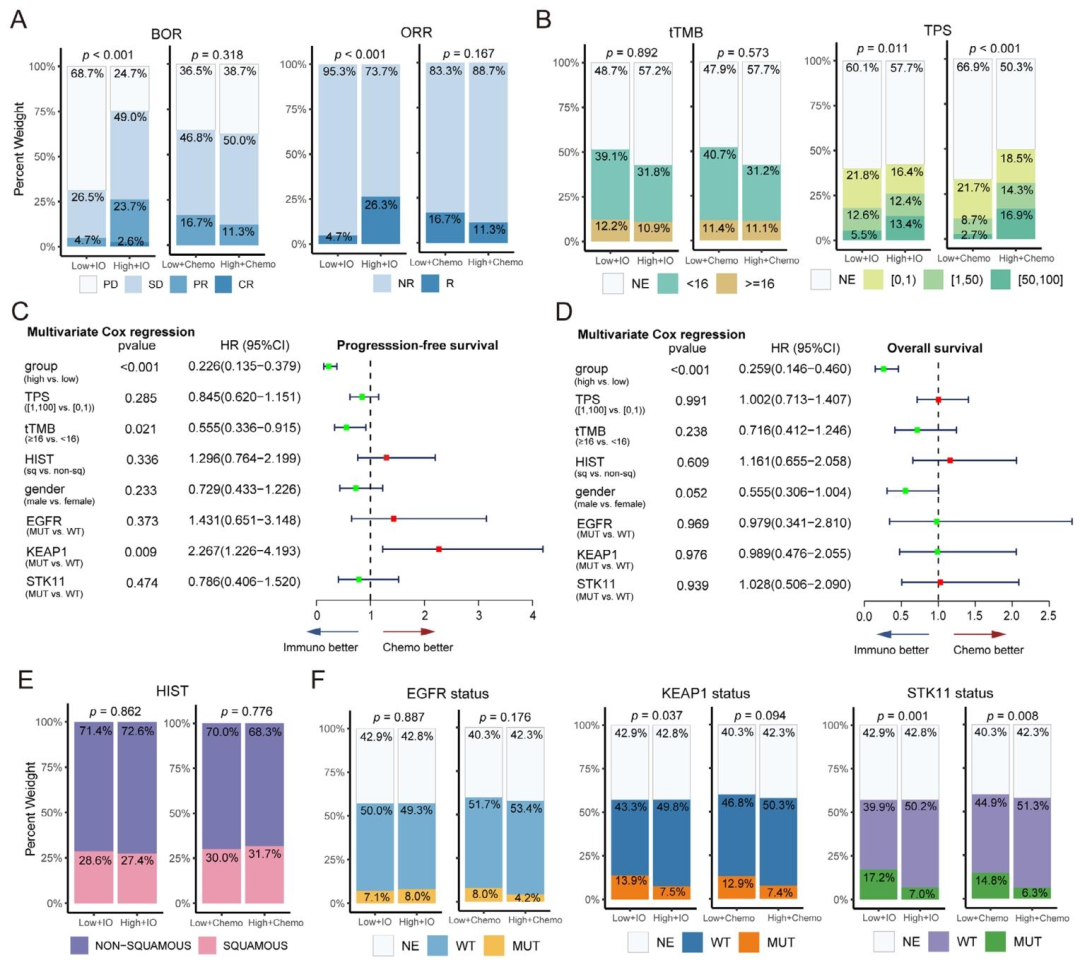
图 3(LIRA与临床特征、突变状态的关联) :图中A-B显示高LIRA评分组有更高的客观缓解率和PD-L1表达;C-D多因素森林图证实LIRA评分独立于TPS、TMB、组织学类型和性别(HR=0.258);E-F显示低LIRA评分组富集STK11和KEAP1突变。影像组学可借鉴:类似地,可将影像标签与关键驱动基因突变状态关联,证明其反映特定生物学背景。
5. 单样本参考数据库实现个体化预测
利用OAK和POPLAR中439例atezolizumab治疗样本建立参考数据库,可对单个新样本进行批次校正和LIRA评分计算。四个案例验证显示,LIRA成功识别出PD‑L1阴性但仍获益的患者及PD‑L1高表达却无效的患者。
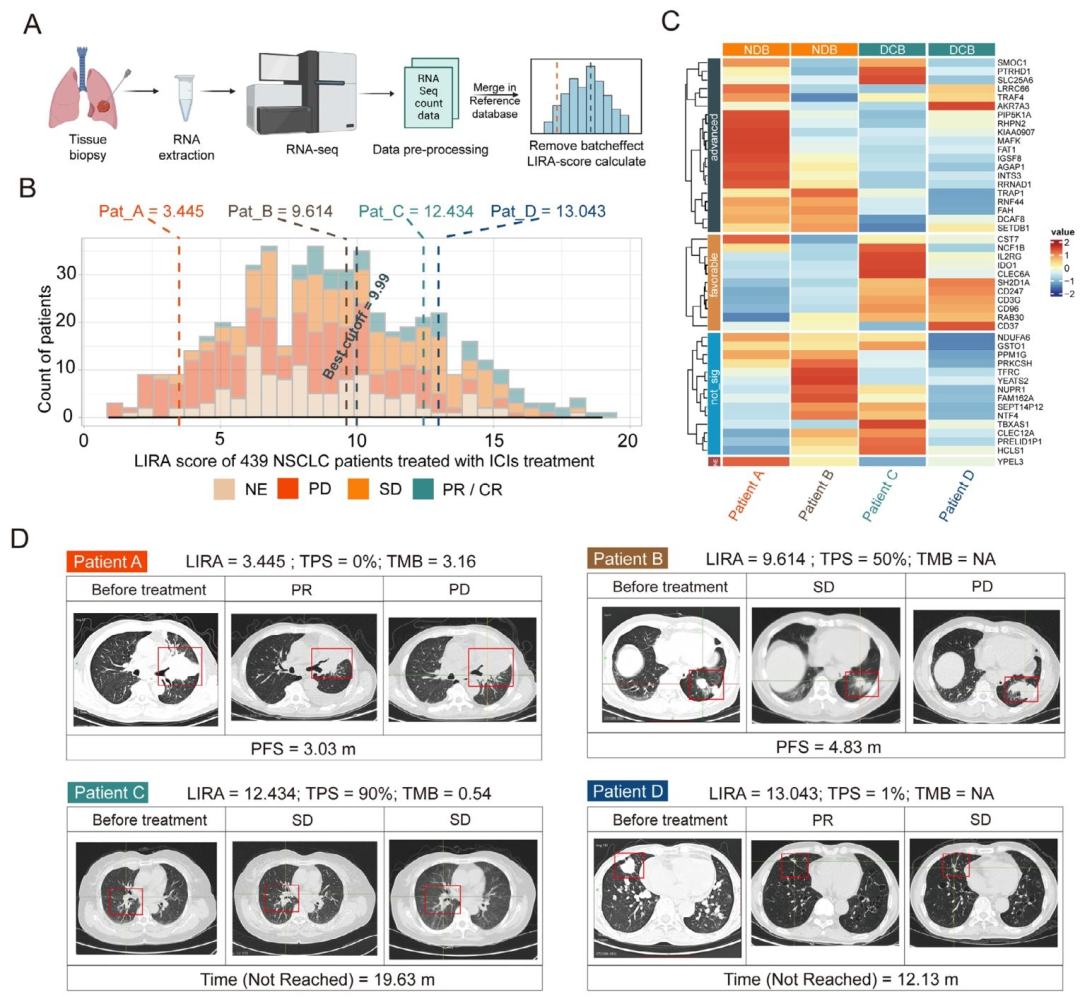
图 4(单样本预测案例) :该图展示了利用参考数据库对四个NFH队列患者进行个体化预测。A为计算流程;B为LIRA评分分布及最佳截断值(9.99);C为模型基因表达热图,低分患者高表达MAFK、FAT1等不良预后基因,高分患者高表达IDO1、CD96等免疫相关基因;D为CT影像,显示低分患者肿瘤进展、高分患者退缩。影像组学可借鉴:通过典型病例展示影像标签如何区分PD‑L1阴性但仍获益的患者,并关联具体基因表达模式。
6. 低LIRA评分富集乙酰化通路及免疫抑制关键基因
差异表达和GSEA分析发现,低分组显著富集组蛋白乙酰化/去乙酰化通路 。HDAC4高表达与不良预后相关(HR=1.48),LRP8‑APOE信号轴在低分组中增强,提示其参与免疫抑制微环境形成。
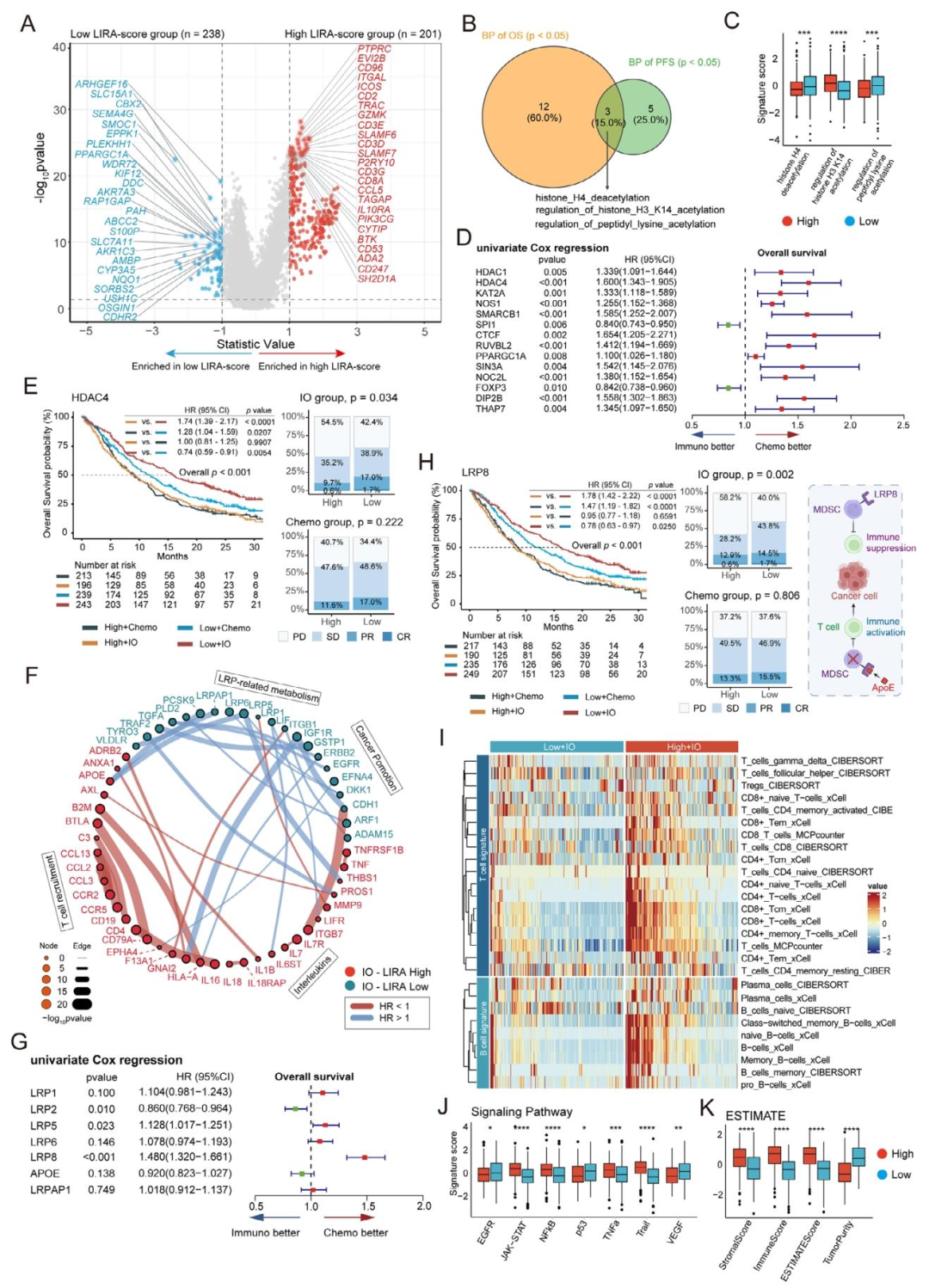
图 5(转录组与代谢特征分析) :A为火山图,显示低LIRA评分组高表达HDAC4、LRP8等基因;B-C为乙酰化相关通路维恩图和ssGSEA评分,低分组显著富集组蛋白去乙酰化通路 ;D-E显示HDAC4高表达与不良预后相关;F-H为配体-受体分析,LRP8-APOE信号轴在低分组中增强 ;I-K显示低分组肿瘤纯度更高、免疫浸润更低。影像组学可借鉴:通过通路富集和配体-受体分析,将影像评分与具体分子通路(如乙酰化、代谢重编程)挂钩。
7. 单细胞测序揭示高/低LIRA组的细胞组成差异
对41,799个细胞的分析显示,高LIRA组T细胞和B细胞比例显著升高 ;低LIRA组则恶性上皮细胞和Treg细胞增多,免疫浸润减少,从细胞层面解释了低评分患者免疫治疗耐药的原因。
8. 深度学习病理模型定位关键组织学特征
基于CLAM框架的LIRA分类器在WSI上生成注意力热图。高LIRA组的高关注区域主要为淋巴细胞聚集区 ;低LIRA组则以肿瘤细胞和成纤维细胞为主,从组织形态学层面印证了分子与细胞水平的发现。
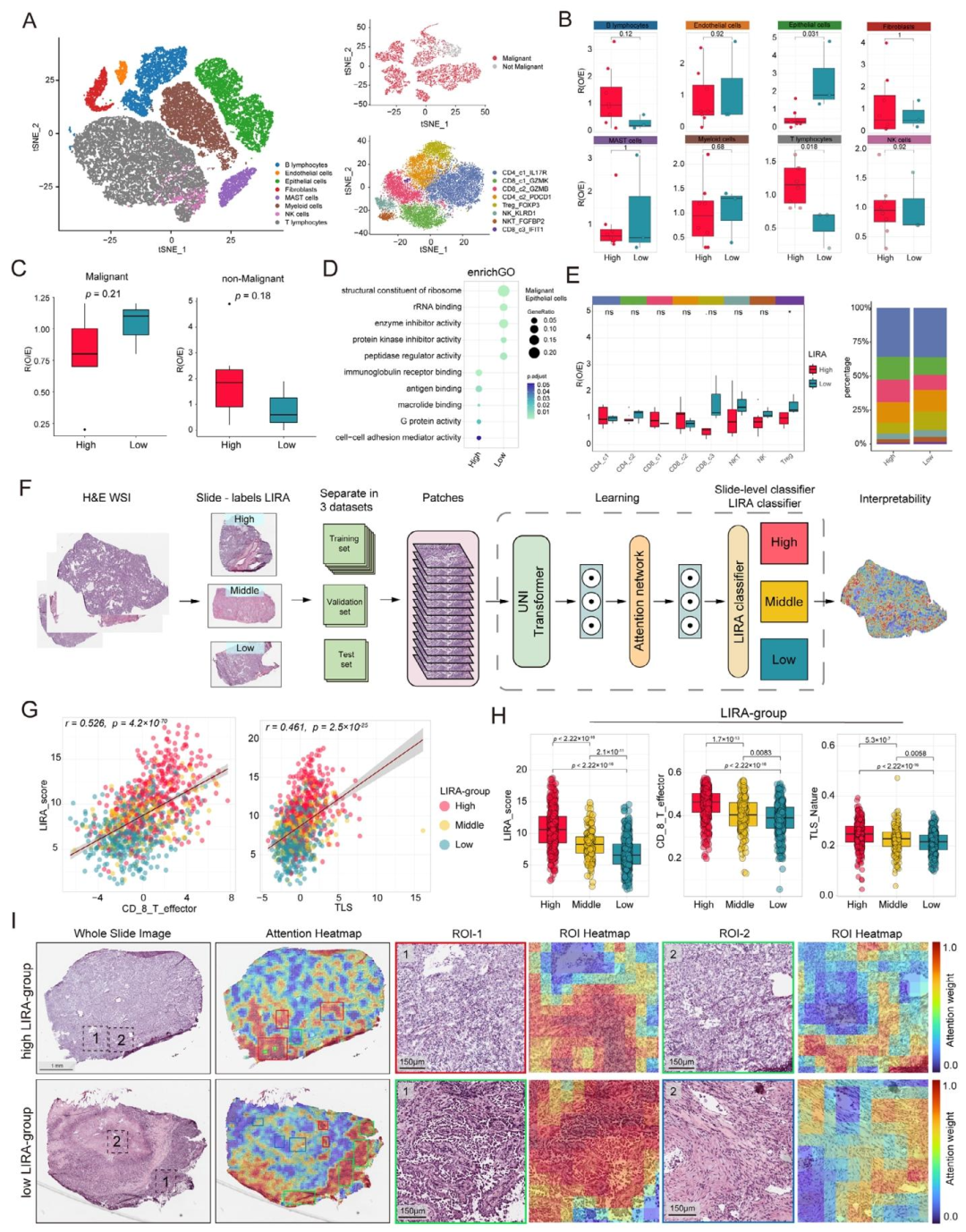
图 6(单细胞免疫图谱与深度学习病理模型) :A为t-SNE图展示41,799个细胞的八大细胞类型;B-C显示高LIRA组T细胞和B细胞比例更高,低LIRA组恶性上皮细胞和Treg细胞更多;D为上皮细胞通路富集分析;E为Treg比例;F为基于CLAM的深度学习模型流程;G-H显示模型预测的LIRA分组与CD8+ T细胞评分、TLS评分呈正相关;I为注意力热图,高LIRA组高关注区域为淋巴细胞,低LIRA组为肿瘤细胞和成纤维细胞 。影像组学可借鉴:通过深度学习热图和单细胞数据,将影像标签的预测决策区域与特定细胞类型(淋巴细胞 vs. 肿瘤/成纤维细胞)直接关联,实现"影像-细胞-机制"三重验证。
07
讨论(把机制故事讲圆)
本研究通过整合多中心随机临床试验的转录组数据,成功构建了名为LIRA的机器学习预测模型 ,并在多个独立队列中验证了其对晚期NSCLC患者免疫治疗生存获益的优异预测能力。与传统生物标志物PD‑L1 TPS和TMB相比,LIRA的AUC提升显著 ,尤其在预测无进展生存期方面表现突出(AUC最高达0.96)。更重要的是,本研究并未止步于模型性能的展示,而是系统性地将LIRA评分与具体的生物学机制挂钩 ,形成了一条从分子到细胞再到组织的完整证据链。机制分析揭示,低LIRA评分并非简单等同于"低免疫活性" ,而是一种由组蛋白去乙酰化异常(尤其是HDAC4高表达)和LRP8‑APOE信号轴激活 所驱动的免疫抑制微环境状态。单细胞数据进一步证实,低LIRA评分肿瘤中Treg细胞比例升高、恶性上皮细胞增多 ,而高LIRA评分肿瘤则以效应T细胞和B细胞浸润为主。深度学习病理模型从组织学层面发现,低LIRA评分的高关注区域主要由肿瘤细胞和成纤维细胞构成 ,这种细胞互作可能物理性阻碍T细胞浸润,从而导致免疫治疗耐药。这些发现不仅解释了LIRA的预测能力,也为逆转免疫耐药提供了潜在靶点(如HDAC抑制剂联合ICI)。本研究的局限性 包括:模型基于回顾性数据,存在选择偏倚;部分队列缺少TPS/TMB数据,限制了与现有标志物的直接比较;未整合多组学数据(如蛋白质组、代谢组);缺乏前瞻性临床验证。尽管如此,LIRA作为一种可解释的、基于转录组的预测工具,有望在临床决策中辅助识别免疫治疗的优势与劣势人群,推动精准免疫治疗的落地。
08
这篇文献的可借鉴思路
这篇论文对影像组学研究最具启发性的核心思路在于:将机器学习的预测评分从"黑箱指标"转化为"可解释的生物学故事" 。传统影像组学论文往往止步于构建一个高AUC的影像标签,却很少回答"这个标签究竟反映了什么生物学过程"。本文提供了一套可迁移的"机制挂靠"方法论 ,影像组学研究者完全可以参照执行。第一,从机制出发筛选特征 。本文并非盲目选用全部差异基因,而是通过"与治疗交互作用显著"这一条件筛选特征基因,确保模型基因与免疫治疗结局存在生物学关联。影像组学同样可以先锚定感兴趣的生物学通路 (如缺氧、糖酵解、乙酰化、T细胞浸润),然后从影像中提取与该通路相关的放射学特征(如纹理、形状、小波特征),构建"通路特异性影像标签"。第二,多组学联合验证机制 。本文利用差异表达、GSEA、ssGSEA将LIRA评分与特定通路(乙酰化)挂钩,再通过Cox回归和KM分析锁定具体基因(HDAC4、LRP8)。影像组学研究可将影像评分与同一批样本的转录组/蛋白组数据进行相关性分析 ,识别与影像评分显著相关的通路和基因,从而赋予影像特征以生物学注释。第三,单细胞数据辅助细胞层面解释 。本文借助公共单细胞数据集,验证了高/低LIRA评分组的细胞组成差异(T细胞 vs. Treg/恶性细胞)。影像组学可与空间转录组或多重免疫荧光结合 ,将影像评分映射到特定细胞类型的空间分布上,例如验证"高影像评分区域是否富集CD8+ T细胞"。第四,深度学习热图提供组织学证据 。本文使用CLAM模型生成注意力热图,并由病理专家标注高贡献区域的细胞类型。影像组学完全可以采用同类可解释性AI技术 (如Grad-CAM、Attention heatmap),将模型决策的区域可视化,并邀请病理医生解读这些区域的细胞构成(如肿瘤细胞、间质、坏死、淋巴细胞簇),形成"影像特征-病理形态-细胞类型"的证据链。第五,构建"评分-通路-细胞-组织"的四层证据体系 。影像组学论文不应仅报告AUC和校准曲线,而应按照本文的范式,依次展示:①影像评分能区分预后;②评分与特定通路(如EMT、代谢)显著相关;③单细胞或免疫组化验证相关细胞类型的丰度差异;④深度学习热图定位关键病理区域。最后,建立单样本预测参考数据库 。本文通过构建参考数据库实现了对新样本的批次校正和个体化预测,这一思路对多中心影像组学研究尤为重要------可建立标准化的影像特征参考数据库,消除不同扫描设备、参数带来的批次效应,提升模型的临床泛化能力。总之,本文最大的示范意义在于:一个预测模型的价值,不仅在于它"准不准",更在于它"为什么准"。影像组学若能将"黑箱标签"转化为"生物标志物",将大幅提升研究的学术深度和临床转化潜力。
09
结语
总而言之,做影像组学不只是拼AUC、堆模型,更要学会给特征找意义、给模型讲道理 。这篇文献给我们的最大启发是:预测评分必须锚定具体的分子通路(如去乙酰化)、关键基因(如HDAC4)、细胞组成(Treg vs. CD8+ T)和组织形态(淋巴细胞区 vs. 肿瘤-成纤维区) ,形成从宏观影像到微观机制的完整证据链 。只有把影像标签与肿瘤微环境、表观遗传、代谢重编程 等生物学过程真正打通,我们的研究才不是"玄学算命",而是有根有据、有血有肉、能讲好疾病故事的真科研 。希望大家以后都能少走弯路,轻松写出有机制、有深度、能发高分的好文章!
参考文献:Wang Z, Fang Y, Huang X, Ma G, Mao Q, Lu X, Rong G, Yu Y, Wang Y, Huang Z, Sun H, Wu J, Gu W, Huang N, Wu J, Zhou R, Rong X, Zheng S, Li S, Wang G, Wang L, Qiu W, Jiang L, Luo P, Liu Y, Bin J, Liao Y, Shi M, Wu Z, Wang J, Liao W, Chen G, Zeng D. Predicting Immunotherapy Outcomes in NSCLC Using RNA and Pathology from Multicenter Clinical Trials. Adv Sci (Weinh). 2026 Jan;13(3):e02037. doi: 10.1002/advs.202502037.