摘要
质谱蛋白质组学可生成表征生物样品中肽段/蛋白质组分的复杂数据,各类机器学习是串联质谱肽段鉴定及数据分析全流程的核心计算方法。随着深度学习成为数据建模与解析的强力机器学习手段,蛋白质组学计算研究者利用海量公开数据集训练机器学习模型,用于预测肽段碎裂谱与液相色谱保留时间。ProteomicsML等资源为这类学习任务提供了详尽的演示教程,缩小了蛋白质组学与机器学习领域的隔阂。但现有深度学习教学材料普遍缺失数据预处理嵌入这一关键步骤:肽段文本序列必须转换为数值格式(即嵌入)才能用于模型训练。肽段嵌入方法种类繁多,性能差异悬殊,但其构建流程与选型依据在蛋白质组学文献中极少被讨论。本技术笔记推出4个谷歌Colab笔记本教程,讲解5种肽段嵌入策略(从简单单数值编码到前沿预训练嵌入),配套代码示例与文字说明,最终教程对5种嵌入方法开展头对头基准测试。教程免费开源,旨在降低研究者将现代深度学习应用于蛋白质组学流程的门槛。
#机器学习 #蛋白质组学人工智能 #教程 #蛋白质组学教育 #肽段 #嵌入 #编码
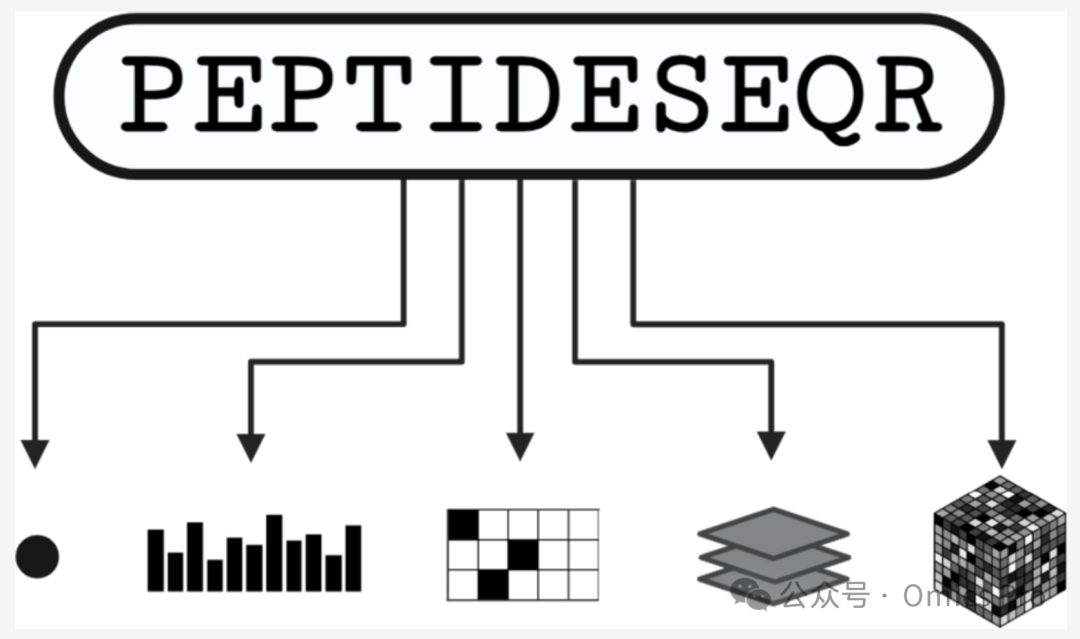
结果
肽段嵌入方法概述
方框1 用于机器学习的肽段嵌入复杂度演进
肽段信息嵌入的替代方法
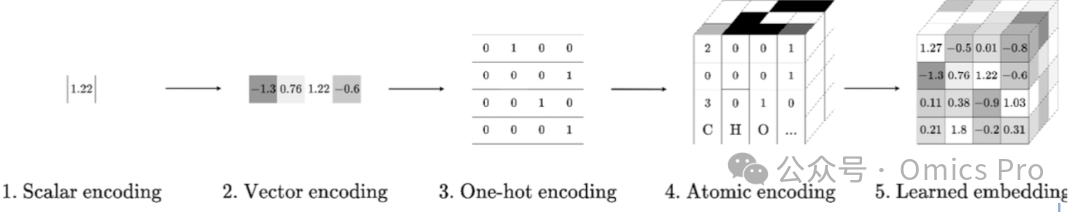
❶ 单一数值(标量):如等电点、保留系数
❷ 1维数值列表(数组/向量):如肽段氨基酸相对组成
❸ 2维数值网格(矩阵):最常见为表征肽段内氨基酸位置的独热编码
❹ 3维人工特征块(张量):如文献中的原子组成编码
❺ 3维学习特征块(张量):数值完全由机器学习生成,实用但难解释
模型训练性能评估
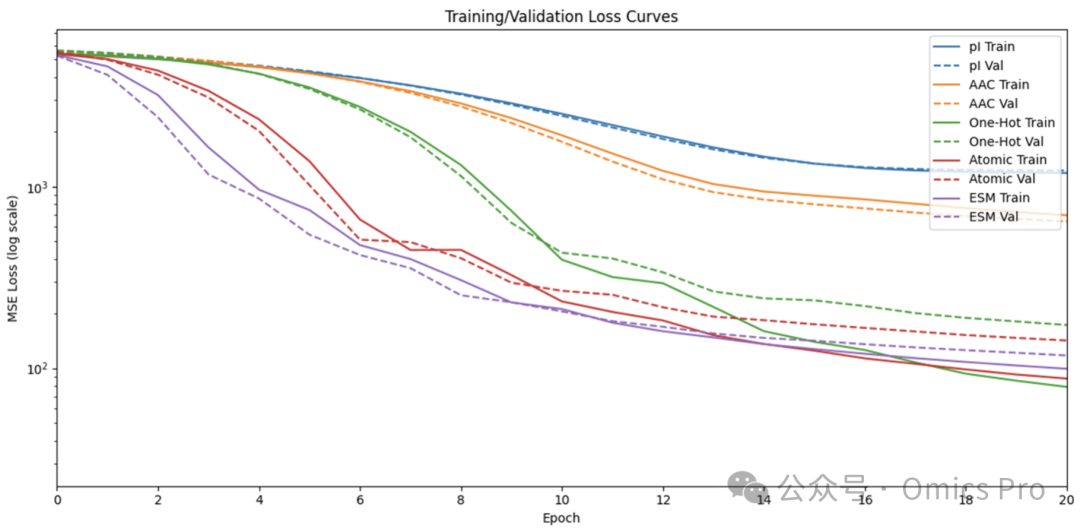
图 1 训练过程中的模型损失
每轮训练后,通过损失函数评估模型在训练集与验证集上的精度,损失值越小模型精度越高;为清晰展示仅绘制20个epoch,完整训练超50个epoch直至验证损失不再下降。
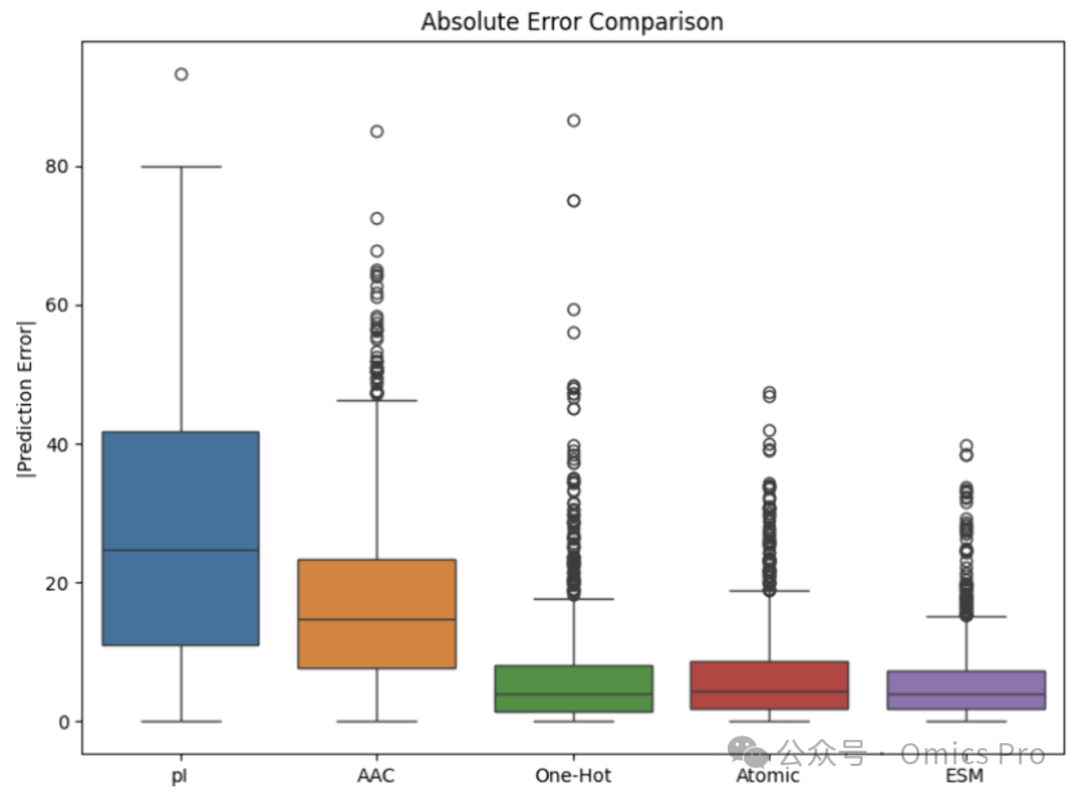
图 2 完全训练模型的保留时间预测误差
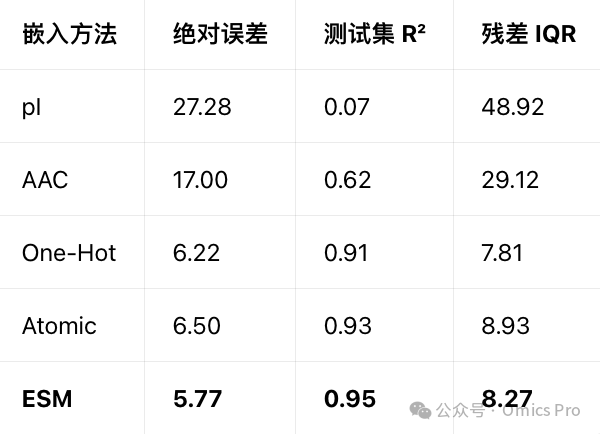
误差为测试集1477条肽段的实测与预测保留时间差值;基于ESM肽段嵌入训练的模型表现最优,中位误差最低、4分位距最小。
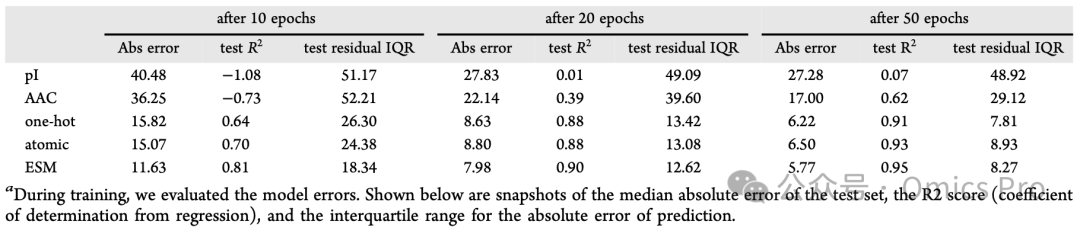
表1 训练过程中的模型性能变化
训练期间评估模型误差,展示测试集中位绝对误差、决定系数(R²)、预测绝对误差的4分位距(IQR)。
数据
https://github.com/PayneLab/ProteomicsEducation
详细总结
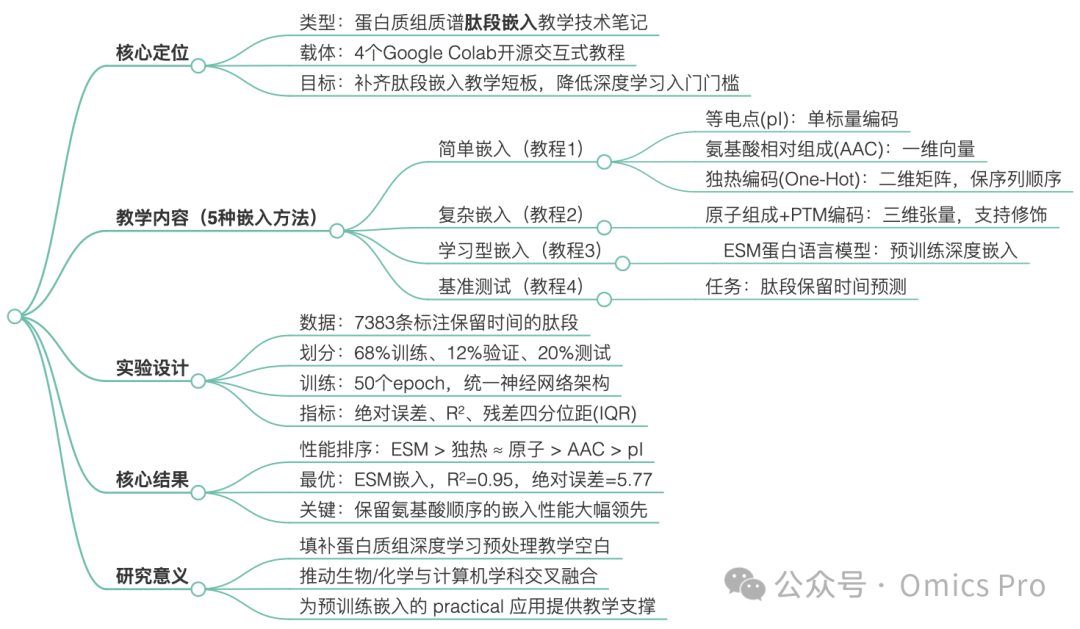
思维导图
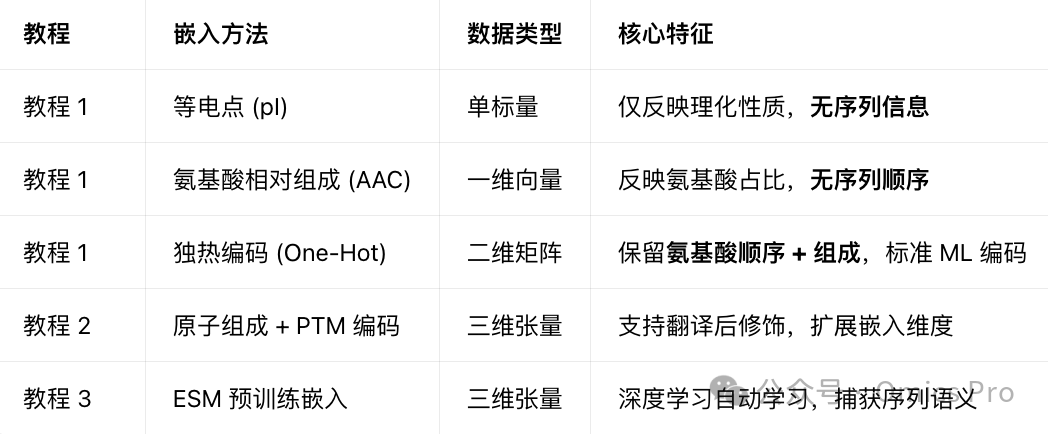
5种肽段嵌入方法(复杂度递增)
测试集最终性能(关键量化指标)
参考
J Proteome Res. 2026 Feb 6;25(2):1160-1165. doi: 10.1021/acs.jproteome.5c00563.
Better Inputs, Better Learning: A Peptide Embedding Tutorial for Proteomic Mass Spectrometry
注:AI辅助创作,如有错误欢迎指出。内容仅供参考,不构成任何建议。