宏基因组学彻底改变了研究人员对微生物群落的认识,微生物组不仅是环境组分,更作为共生体深刻影响着宿主的健康与功能。
鉴于微生物群落固有的复杂性及其所处环境的多样性,研究者进行一些宏基因组学研究时必须精心设计以获取能真实反映目标群体特征的准确结果。来自美国加州大学等机构的研究者在 Nature Reviews Methods Primers IF: 50.1 上发表了一篇宏基因组数据分析入门指南,该指南系统梳理了宏基因组学的方法学进展与现行实践:从样本采集、DNA提取的初始阶段,到用于数据分析的先进生物信息学工具,重点阐释下一代测序技术对研究规模与精度的深远影响。

▲ DOI: 10.1038/s43586-024-00376-6。
作者批判性评估了宏基因组实验体系、现有技术与计算分析方法中存在的挑战与局限。除技术方法外,它们还探讨了宏基因组在人类健康、农业与环境监测等领域的应用前景。

▲ 研究团队。
0.引言
宏基因组学是一门融合实验与计算方法的交叉学科,致力于解析微生物群落的基因组组成与功能作用。典型的宏基因组研究始于目标环境样本(如土壤、水体、血液或粪便)的采集,随后提取样本总 DNA 并通过测序技术获取基因组位点的测序读段,最终构建宏基因组图谱以全面解析特定样本微生物组成。该方法区别于基于扩增子的细菌分析策略,后者通过选择性扩增并测序 16S 和 18S 小亚基核糖体 RNA(SSU rRNA)特定区间。简而言之,就是看看特定样本中的碱基序列来自于哪些微生物群体,进而分析不同群体的丰度与功能。
宏基因组学最初作为探索土壤、水体等环境样本中庞大微生物多样性的强效工具,用于揭示自然环境中微生物群落的复杂性与功能。随着研究人员的持续使用与一系列工具的开发,宏基因组学已成为微生物群落研究的基石方法,推动人类健康、食品安全、农业与生物技术等关键领域的进步。
早期的宏基因组技术依赖 桑格测序(一代测序),需将随机片段化的DNA克隆到可培养细菌中。而当前最流行的方法则以下一代测序技术(NGS)为核心,主要是 illumina 公司开发的桥式短读长高通量并行测序方法,使单碱基测序成本降低数个数量级,大幅提升了宏基因组数据产出的可能性(图1)。测序成本的优化催生了多项宏基因组项目,这些项目对发现新微生物、深化理解微生物与环境互作机制至关重要。
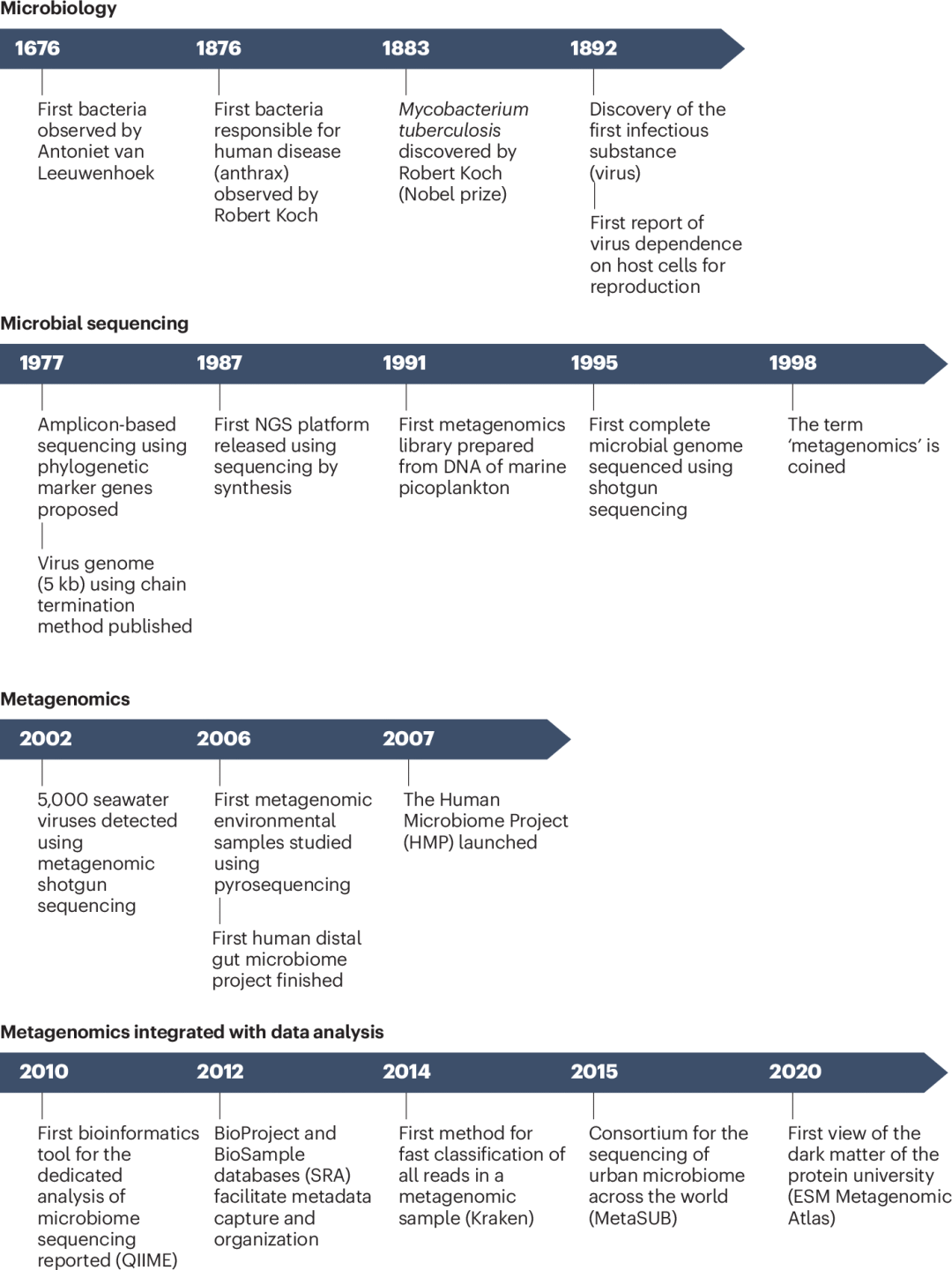
▲ 图1:微生物发现与宏基因组分析技术发展历程。从早期微生物学发现到测序技术进步,宏基因组学领域已逐步产生专注于微生物与宏基因组发现的数据库、工具及组织体系。
尽管宏基因组研究取得显著进展,样本处理标准化协议的缺失仍制约结果的可重复性,即样本采集与 DNA 提取方法显著影响 DNA 质量与产量。在环境宏基因组学研究中,污染问题尤为突出,尤其对低生物量样本或受极端气候等环境因素干扰的样本。测序技术的选择进一步限制样本制备流程。此外,参考数据库缺乏统一标准导致记录不完整且不一致。上述待解决的问题使得优化实验流程、计算方法与分析策略仍是当前的研究热点,研究人员正致力于标准化多样本类型的采集与处理流程,以降低污染风险、解决低生物量样本难题并提升分析稳健性。
1.宏基因组实验指南
选择合适的样本采集方法、保存技术与测序流程,是构建可靠下游宏基因组预处理与分析的基础。由于每个步骤均可能引入偏倚,从采样到实验室分析的完整流程需精心设计与执行。根据微生物组的样本来源(宿主相关或环境样本),需采用不同的标准化方案。采集后的样本在运输与储存前需谨慎处理,以维持DNA提取前宏基因组信息的完整性。DNA提取后需进行文库构建,包括DNA片段化、末端修复、接头连接与序列索引标记等步骤,使样本DNA适配测序平台要求。下文详述宏基因组样本实验流程(实验流程概览见图2)。
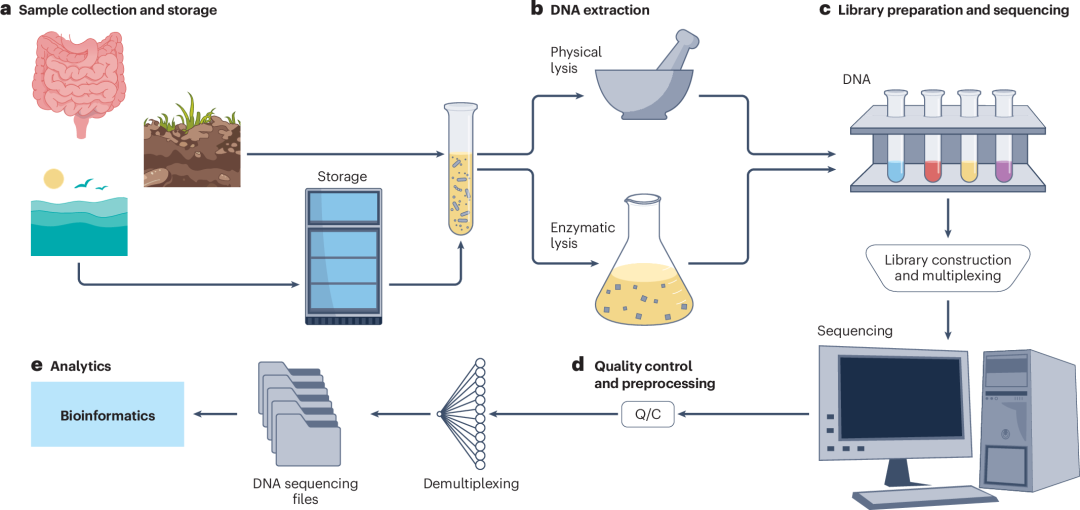
▲ 图2:宏基因组实验流程。
上图所示流程按顺序如下:
-
a. 样本收集以后立即保存或处理(如土壤/水体环境样本或宿主微生物组样本);
-
b. 物理或酶法裂解提取DNA;
-
c. 构建DNA文库,通过DNA条形码标记实现多样本混合测序(multiplexed),随后进行测序;
-
d. 测序读段进行质量控制与预处理;
-
e, 通过条形码 barcode 拆分不同样本来源的序列,随后经生物信息学分析处理。
研究设计
严谨的研究设计可最大化生成高质量、可重复数据的可能,并确保结论的科学性与可靠性。研究者应根据研究目标(人类、动物或环境研究)选择最适设计类型:
-
观察性研究:通过无干预监测揭示微生物组的自然变异;
-
横断面研究:提供单时间点的微生物组快照,用于发现微生物组与特定结局(如健康状态)的关联;
-
病例对照研究:比较患病与健康个体的微生物组差异;
-
纵向研究:追踪微生物组随时间变化,解析时间动态特征;
-
随机对照试验:作为因果推断的金标准,通过最小化偏倚评估干预措施效应。
确定研究设计后,务必需通过功效分析(基于预实验或文献效应值估计)确定样本量以确保统计效力。
微生物组样本采集
样本采集需考虑以下几点:样本是否代表研究目标、是否存在外源污染或交叉污染风险、采集成本与效率等实操性因素,以及阴阳对照设置。对于不同来源的样本其注意点也有所差异:
-
人类微生物组样本:已知母乳、肿瘤、呼吸道、阴道环境、泌尿道、皮肤与唾液等多种样本中存在微生物群体,但粪便样本因高生物量与易获取性仍是肠道微生物组研究的主要材料。采集时需平衡患者舒适度与微生物群落保存效果。住院与门诊样本采集的会遇到的相同问题包括:①样本保存冷冻条件、②污染防控、③低生物量样本处理与④医疗资源可及性。针对粪便样本,尽管不同保存方法对 DNA 组成影响存在争议,但新鲜样本速冻仍被视为获取可靠分类学分辨率的金标准。商用居家粪便采集推荐采集晨便并立即冷冻或保存于稳定溶液;
-
非人类动物样本:动物研究中,采样策略取决于研究目标、目标组织与宿主物种。肠道微生物组研究可采用粪便样本、直肠拭子或尸检肠道内容物;
-
环境样本:①环境宏基因组样本包括空气、物体表面、水体与土壤。空气样本通常通过滤膜富集微生物,但低生物量与温湿度等气象因素会影响微生物群落;②城市高频接触表面样本可用植绒拭子采集,其微生物组成受表面类型、清洁频率与人类活动影响;③水体样本需在预设深度采集并现场过滤;④土壤样本需使用灭菌工具采集并保存于无菌袋,需关注土壤表层与深层微生物密度差异导致的群落特征变化。作者建议多时间点与多地点采样以捕捉自然波动,结合环境元数据(温湿度、营养浓度等)校正混淆因素确保差异反映真实生物学或环境变异。推荐采用一致性采样策略降低污染,并采集阴性对照(如采样设备拭子、邻近空气样本等)。
样本处理、运输与储存
微生物群落具有代谢活性,采集后需立即冷冻灭活并稳定保存至DNA提取。温度波动、氧气暴露与反复冻融会破坏核酸完整性,影响宏基因组图谱,且冻融循环可能筛选出难裂解细菌引入偏倚。
无菌采集后液氮速冻并-80°C保存为金标准,但野外研究常需替代方案。评估过的稳定方法包括RNAlater、EDTA、乙醇或异硫氰酸胍基稳定剂。使用稳定剂时需保持适当比例并充分混匀。稳定剂选择、储存时间与温度均影响样本降解,甚至-80°C保存样本亦可能因稳定剂效率差异改变分类组成(如粪便样本)。长期储存或温度波动可损害微生物完整性(如猪粪与污水样本),故优化保存方法对结果可靠性至关重要。
建议在元数据中记录保存方法与温度,并将信息上传至protocols.io与STAR methods等平台。长期冻存前分装避免冻融循环,并设置阴阳对照以量化潜在偏倚。尽管保存方法偏倚难以完全消除,但优化储存条件与生信流程可最大限度降低其对研究目标的影响。
DNA提取
DNA提取(图2b)涉及微生物裂解与核酸纯化。不同裂解方法具有独特偏倚,导致该步骤成为样本制备中变异最大环节。方法选择取决于测序平台(短读长或长读长):短读长测序需珠磨破碎法有效裂解革兰氏阳性菌与真菌,减少手工操作时间;长读长测序则采用酶解法避免机械裂解导致的DNA断裂(但真菌酶解仍存挑战)。近年来长读长DNA提取技术进步催生新方案,各种技术蓬勃发展。苯酚-氯仿法、CTAB法等提取试剂盒通过不同技术提高产量并去除抑制剂,但尚无单一方法适用于所有环境样本。
设置阴性对照(如分子级水空白样本)可识别采样与提取阶段污染;阳性对照(如人工菌群)或内标(外源DNA/RNA)有助于量化浓度偏差、提取效率与生信分析偏倚。
文库构建与测序
DNA提取后,文库构建(图2c)将DNA转化为标记文库以适配测序平台。短读长文库构建包括四步:超声或酶切随机片段化DNA、末端修复、连接双端或单端接头、索引标记。索引标记支持多样本混合测序),降低成本并提升通量。
Ⅰ 测序平台
-
Illumina NGS平台:因高通量、高精度短读长(50-300 bp)成为宏基因组研究主流,适用于变异检测、基因表达分析与宏基因组图谱构建。
-
Nanopore(ONT)与 PacBio 等长读长技术:可生成数百kb读长,支持实时数据采集与复杂区域解析。其直接测序天然DNA避免PCR偏倚,保留表观修饰,在从头组装与异构体鉴定中优势显著。例如,长读长可精准解析抗生素耐药基因的遗传背景及其与可移动元件或宿主的关联,减少短读长偏倚。但 Nanopore 平均碱基错误率(4-10%)高于Illumina(约0.1%),且读长产量较低。尽管如此,PromethION平台、R10.4.1 芯片与 PacBio Sequel II 系统的 HiFi 测序等技术正逐步改善覆盖深度与错误率问题。
选择平台需权衡研究目标、样本基因组复杂度及成本效益。长读长测序成本显著高于短读长(单位数据量成本高数倍),主要源于运行时长与文库复杂度。多样本混合测序策略结合短读长经济性与长读长连续性,成为高质量基因组组装的经济方案。
Ⅱ 文库构建与测序
Illumina文库分为 PCR 扩增与非扩增法:后者需至少25ng DNA输入,前者灵活性高但可能引入扩增偏倚。文库构建后,带接头的DNA分子通过桥式扩增固定于流动池表面进行光学测序,支持单端与双端测序。
长读长测序效果高度依赖文库制备。过度片段化会损害读长与质量。ONT DNA连接法16S 建库试剂盒中,DNA 剪切至>8 kb片段,随后经过末端修复后连接蛋白偶联接头并进行预处理;PacBio SMRTbell 建库通过连接通用发卡接头适配高精度长读长测序。纳米孔与PacBio单分子实时测序的优势在于跨越复杂基因组区域。纳米孔通过电流波动识别碱基乃至其修饰状态;PacBio提供高精度 HiFi 读长(环状共识测序)与超长读长模式(半数读长>50 kb。二者读长显著优于Illumina,但存在碱基识别上下文依赖性错误(纳米孔)或需多次识别提高精度(PacBio)的局限。
基于染色体构象捕获(Hi-C)的配对读长技术通过连接物理邻近DNA区域,可跨越广泛基因组区域。宏基因组Hi-C(metaHi-C)结合鸟枪测序与Hi-C,将同一细胞的邻近DNA连接,辅助contig组装。
2.宏基因组结果分析
宏基因组数据分析的初始阶段包括数据预处理和质量控制步骤,具体涵盖 read 覆盖度评估、基于参考基因组的测序深度分析、重叠群完整性评价以及组装重叠群中污染水平的额外测定。随后的生物信息学分析将转向计算需求更高的任务,如宏基因组组装和微生物群落功能分析,这些步骤需要高性能计算集群等先进基础设施以实现高效数据处理。接下来将系统探讨宏基因组数据处理与分析中常用的一系列策略和生物信息学工具,并讨论有效获取可靠结果所面临的挑战及其缓解方案。
Ⅰ 质量控制
在质量控制前,解复用步骤(demultiplexing)基于文库构建时连接 read 上的特异性索引区分个体样本。目前已开发多种解复用工具,包括Flexbar、Ultraplex 及其他工具。
宏基因组样本的全基因组鸟枪法测序具有随机性特征,可能在进行宏基因组组装时引入偏差,从而影响样本内基因组的精准重建。因此,解复用后对原始测序数据进行质量评估尤为重要。read 即读长的质量通过测序平台提供的 Phred 质量分数衡量,该分数基于碱基错误测序的概率计算。低质量读长(Phred分数<30)通常含有技术伪影,如测序错误、PCR产物残留和接头序列,需使用 FastQC、PRINSEQ、Trimmomatic、BBTOOLS等质量控制与数据清理工具予以剔除。这些工具通过整合测序仪器提供的质量信息和包含接头/引物序列的数据库,实现测序接头序列的去除,并检测读长序列中的异常现象(如可能由于污染或基因组重复影响的k-mer过度富集现象,此类重复会导致复杂的宏基因组组装)。此外,这些工具还可评估读长长度、质量分数、GC含量及模糊碱基数量/百分比。经修剪后低于预设长度阈值的读长将被剔除。
针对长读长测序数据,另有专门的质量控制方法。例如,NanoFilt 工具根据平均读长质量、长度和 GC 含量等常见参数对长读长进行过滤;LongQC 通过评估"无意义读长"(可能源自低质量纳米孔)的比例来评价长读长质量。此外,宿主基因组或其他污染源产生的读长必须被去除,DeconSeq 和 KneadData 等自动化去污染工具可有效简化这一流程。
Ⅱ 宏基因组组装
虽然可直接对宏基因组样本的单个读长进行分析,但将读长组装为单个重叠群和宏基因组组装基因组(MAGs)通常更具优势------这种方法能够以更高分辨率(实现种/株水平更精细的区分)解析微生物群落的基因组组成,并提升功能注释的准确性(图3,左侧)。
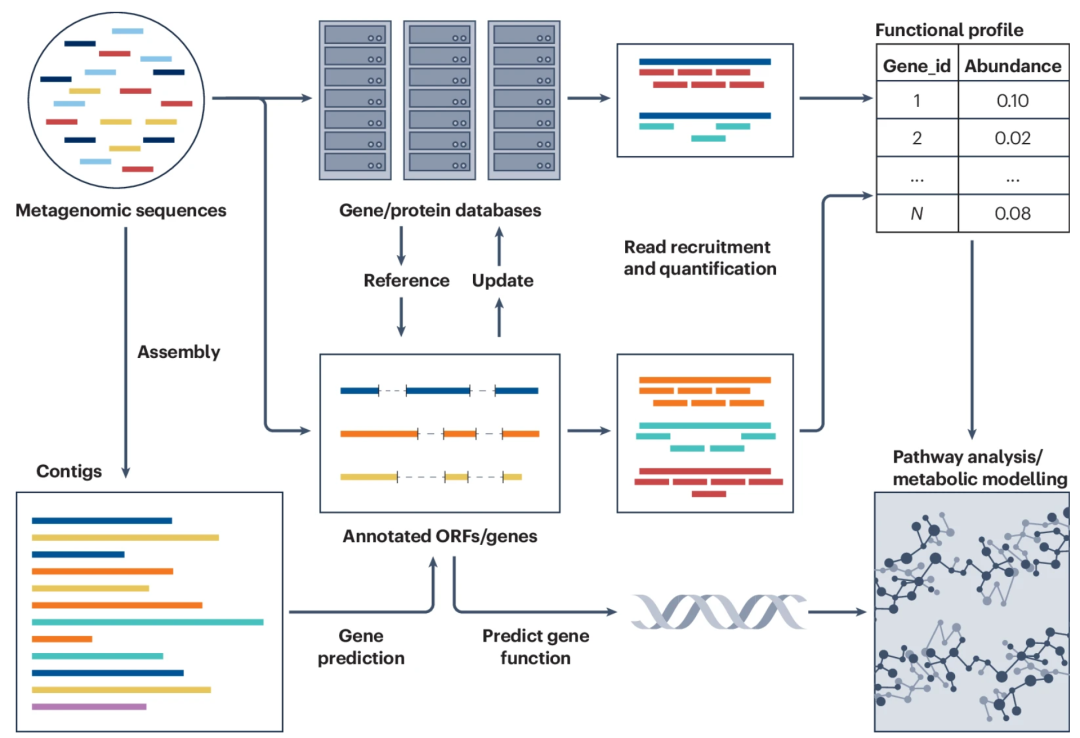
▲ 图3:宏基因组功能分析。宏基因组序列是开展功能谱分析和注释的常规起点(左侧)。已知基因可通过比对参考基因组实现基于比对的定量分析(上行)。宏基因组测序读长也可用于从头基因预测工具训练模型,通过识别输入数据中的开放阅读框(ORF)进而进行读长定量(中行)。基于比对和从头预测的方法均可用于功能谱分析,所得结果可进一步挖掘生物学意义(右侧)。通过将宏基因组序列组装为重叠群,可促进新 ORF 的发现,并基于现有知识预测基因功能(下行)。
(1) 常见的组装方法
宏基因组组装工具主要基于单基因组组装算法的扩展,这些算法经过改良以适应宏基因组数据中普遍存在的不均匀覆盖度及物种/菌株多样性特征。多数短读长宏基因组组装器采用 德布鲁因图(de Bruijn graph)方法,即通过从读长中提取 k-mer 构建组装图谱。
基因组重复序列会导致组装模糊性,从而难以确定与基因组正确重建相对应的路径。在单基因组分析中,可通过分析组装覆盖深度(即每个k-mer在测序读长中的出现次数)识别甚至解析重复序列。短读长宏基因组组装器已扩展至长读长测序技术。例如,针对宏基因组样本的 metaFlye 组装器通过计算局部邻域内的k-mer频率来适应样本内的覆盖度差异,而单基因组长读长组装工具 Flye 则未采用此策略。类似地,hifiasm-meta 通过调整短读长和嵌合读长的剔除标准,对hifiasm工具进行改良,以应对样本中某些基因组可能存在的低覆盖度问题。hifiasm-meta和hifiasm专为 Pacific Biosciences 的HiFi技术开发。另一针对高质量长读长宏基因组数据组装的工具metaMDBG,则以读长中识别的最小化器(minimizers)为核心构建组装。最小化器是一种以低内存占用量高效压缩基因组数据的策略,相较于德布鲁因图方法具有速度和内存优势。
混合组装方法结合短读长的高精度与长读长的广覆盖性,为宏基因组组装提供了高效解决方案。近期一项研究评估了十组环境宏基因组样本及计算机模拟数据,使用七种组装工具(IDBA-UD、MEGAHIT、Canu、metaFlye、Opera-MS、metaSPAdes和hybridSPAdes),结果显示混合组装工具(Opera-MS和hybridSPAdes)在准确性方面持续优于其他工具。混合组装基准测试表明,Unicycler 在生成连续基因组方面优于 MaSuRCA 和 SPAdes,尤其在结合 Illumina 与 ONT 数据时表现突出。
尽管混合组装技术有所突破,但利用高错误率长读长重建复杂宏基因组(如污泥或废水样本)的高质量基因组仍面临挑战。为此,一种新型单倍型解析分层聚类混合组装(HCBHA)方法应运而生。该方法通过将短读长与长读长分型为不同单倍型,再分别组装各细菌基因组,从而实现了从高度复杂生态系统中重建近乎完整的基因组。宏基因组样本中可能包含同一物种的多个菌株,各菌株间仅存在少量基因组位点差异。此场景需借助类似于人类基因组单倍型分型工具的专用组装器,以分别组装各菌株的重叠群。与人类基因组仅含两种单倍型不同,样本中基因组变体的数量通常未知且远超两个。
需指出的是,多单倍型问题属于"NP难"问题,其高效求解算法极难存在,现有方法均依赖无法保证结果正确性的启发式策略。此类方法可能遗漏菌株或生成样本中实际不存在的单倍型嵌合体。测序错误及宏基因组数据的片段化、不完整性进一步加剧了挑战。另一替代策略是仅基于保守基因集定义单倍型或菌株,无需高质量组装即可实现。
(2) 重叠群分箱
宏基因组组装器生成的重叠群可能来源于不同生物体,无法预先分离。这些重叠群可通过 分箱(binning)归类为代表单一生物体或分类群的集合,其依据是组装过程中未利用的特征信息。通常假设同一样本中覆盖深度相似、覆盖模式一致或k-mer组成相近的重叠群源自同一基因组。
宏基因组分箱工具通常通过标注重叠群特征(如覆盖度、序列组成)并结合聚类算法构建分箱。例如,MetaBAT采用改进的k-medoid算法,基于四核苷酸相似性与丰度信息结合的距离度量对重叠群聚类。CONCOCT则在高斯混合模型中整合重叠群丰度与序列组成信息以识别聚类分箱。最新工具VAMB依赖变分自编码器(一种神经网络),将初始表示为高维空间点的重叠群聚类,其嵌入空间由跨样本丰度及序列组成定义。MaxBin和Binnacle等工具还额外利用配对末端信息辅助分箱。若分箱结果接近完整基因组重建,则称为宏基因组组装基因组(MAGs),以区别于纯培养分离株的基因组。
Ⅲ 宏基因组分类方法
分类学分类与谱系分析旨在利用已知微生物类群的测序基因组作为参考,识别并估算宏基因组样本中各类微生物群的相对丰度。该方法依赖于目标类群的序列数据,与前述旨在从样本DNA片段中发现新微生物基因组的宏基因组组装策略不同。分类学分类与谱系分析均通过比对测序读长或重叠群与参考数据库实现,但方法与目的存在差异:分类学分类通过"分类分箱"(taxonomic binning)将单个读长或重叠群分配至特定类群,进而聚合这些分配结果以估算样本中各类群的相对丰度;而分类学谱系分析则通过比较样本整体序列内容与参考序列直接报告类群丰度。
① 首先是基于比对的策略。基于比对的方法依赖算法将测序读长与参考基因组进行比对(图4Bb,Bc)。当前此类算法通常采用分类分箱策略,确定每个读长在参考基因组上的潜在位置,评估比对质量得分,并基于与参考基因组的序列相似性为比对读长分配分类学标签(图4Bd)。

▲ 图4:基于比对与无比对策略的宏基因组分类学分析。基于无比对谱系分析工具通常计算负担较低:首先生成参考数据库与宏基因组样本的k-mer谱(图4Aa);随后比对两者k-mer以识别匹配序列(图4Ab);最终量化匹配k-mer生成分类学谱(图4Ac)。基于比对的方法敏感性更高但计算成本更大:宏基因组读长与预建的标记基因参考数据库比对(图4Ba);通过索引、动态规划或k-mer匹配计算读长与基因组的相似性(图4Bb);读长比对至参考数据库(图4Bc)后量化生成分类学谱(图4Bd)。
全基因组比对策略是最早用于分类学分析的算法。例如,MEGAN通过BLAST比对读段,并将其分配至最低共同祖先(LCA)------即读段可能来源物种群的共同祖先节点。LCA 策略的引入是因为宏基因组读段通常较短且碎片化,可能与参考基因组不完全匹配,或由于近缘物种/菌株间基因组高度相似而匹配多个参考基因组。通过确定与查询序列匹配的多个参考序列的 LCA,这种方法可稳健处理不完整或分歧序列,也可用于不依赖比对或混合分类分析(图4Bd)。MEGAN将保守性较低的物种置于系统发育树的"叶端",保守性较高的物种靠近"根部"。尽管MEGAN针对短读段优化,但其分类分箱策略已通过MEGAN-LR134工具适配于长读段及组装重叠群。
② 其次是标记基因优化策略。单拷贝特异基因的精选子集可用作标记基因,提升比对方法的计算效率。基于标记基因的策略仅用标记基因构建参考数据库,以减少数据量并优化资源利用(图4Ba)。例如,RefSeq 数据库从 9.8 TB缩减至 MetaPhlAn 的 10.41 GB,降幅达 98.95%。不同工具使用的标记基因数量差异显著:MetaPhyler 选用31个蛋白编码标记基因(涵盖581属、214科、99目、46纲、27门),而 PhyloSift 则整合37个核心基因家族及 16S/18S rRNA 等补充集,总计约 800个 基因家族。
16S rRNA基因作为细菌和古菌分类的关键标记,其保守区与可变区共存,其中V4区因高变异性常用于扩增子测序,支持物种水平分辨率。MetaPhlAn 等工具通过比对分支特异性标记基因评估微生物丰度,其标记基因选自200余万个候选基因,具备分支内高保守性和分支外低相似性。MetaPhlAn4进一步支持从宏基因组组装基因组(MAG)构建自定义标记基因数据库,结合参考基因组数据提升分类谱的准确性和特异性。
基于操作分类单元(mOTU)的标记基因策略通过成对比对聚类标记基因。目前,已鉴定出10个适用于构建7700余个参考mOTU的基因,并新增来自15万个MAG的2万余个mOTU。标记基因方法因依赖保守序列,特异性(真阴性识别)较高,但灵敏度(真阳性识别)较低。
③ 另外一种是基于k-mer的无比对策略。这种方法通过生成k-mer谱(图4Aa)、比对参考数据库(图4Ab)并定量共享k-mer生成分类谱(图4Ac),计算负担低于比对策略。k-mer策略通过精确匹配、分箱、哈希算法及判别子集等技术实现扩展。例如,Kraken 通过多数投票策略分类读段,但其在低分类层级可能因序列相似性或测序误差表现不佳;Bracken 通过概率重分配读段提升细分层级的准确性。
④ 最后一种策略是混合策略,混合方法结合比对策略的物种/属级高精度与无比对策略的高效性。例如,Metalign 通过MinHash(k-mer子采样估计基因组相似性)结合比对,利用包含指数(样本中参考基因组存在的可能性)优化分类谱分析,既保持精度又提升效率。
在测序方法层面,目前很多文章对短读长与长读长分类工具进行了多种层面的比较。
如前所述,长读长测序技术是指以Pacbio、牛津纳米孔为主的第三代测序技术。与二代测序(即短读长,以Illumina为主)相比,长读长测序技术提供更完整的序列信息,显著提升复杂微生物群落的分类分辨率和准确性。一项评估11种分类方法的研究表明,长读长分类器(如BugSeq、MEGAN-LR)整体优于短读长工具,尤其在低丰度物种检测中表现突出(如PacBio HiFi数据中 0.1% 丰度物种的高精度检测)。长读长数据在物种和属级分类中均提供更优的检测指标,且无需过滤即可实现高精度与召回率。
值得注意的是,读长质量显著影响长读长分类工具性能:短片段(<2 kb)占比过高会降低精度和丰度估计准确性。此外,ONT平台的实时测序能力可直接用于分类,避免了传统方法依赖培养和分子诊断的耗时过程,对临床宏基因组应用中病原体的快速精准鉴定至关重要。
Ⅳ 宏基因组功能分析
Ⅰ 宏基因组功能分析
宏基因组功能分析旨在解析微生物群落在特定环境中的功能潜力及代谢通路,揭示其生化途径、酶活性及生态系统作用。早期研究通过克隆环境DNA功能基因至大肠杆菌并检测酶活性实现非计算分析,后续工作逐步证实环境样本中存在多样微生物功能。大规模人体肠道微生物研究(如人类微生物组计划)表明,微生物功能比物种组成更具跨群体保守性,提示功能群组可能比分类单元更适合跨研究比较。本节将探讨功能分析的关键流程。
Ⅱ 基因预测方法
基因预测通过识别测序读段或组装重叠群中的潜在基因区域(图3下图),核心挑战在于区分真实蛋白编码基因与类基因序列。完整基因组中,基因区域表现为开放阅读框(ORF),而宏基因组数据的片段化需考虑不完整ORF。早期基于比对的工具(如CRITICA、Orpheus)通过比对已知蛋白数据库推断基因存在(图3上图),但仅能检测已知蛋白基因,限制新基因发现。
现代从头预测工具(如MetaGeneAnnotator、Prodigal)利用统计模型(如k-mer频率、ORF长度、GC含量、序列模体)预测基因,并整合隐马尔可夫模型、支持向量机及条件随机场等算法提升准确性。由于不同细菌物种的模型参数差异,宏基因组样本中ORF的物种归属未知,MetaGeneAnnotator通过物种特异性特征(如核糖体结合位点模式、双密码子使用偏好)优化预测精度。Prodigal则基于起始密码子、核糖体结合位点模体及六聚体使用模式构建通用模型。深度学习工具(如CNN-MGP、Meta-MFDL)近期展现出潜力,但尚无统一金标准。
Ⅲ 功能注释
功能注释通过比对未知基因与参考数据库(如eggNOG-mapper、KEGG KOALA),基于同源关系分类基因至功能群组(图3左)。COG、KEGG等数据库支持基于直系同源的分类,而MorF、FunFams等工具结合结构相似性及聚类策略。ProtBERT、ProSE利用序列嵌入技术,Pfam、AGNOSTOS-DB等数据库则通过蛋白家族分类辅助功能边界解析。
传统方法依赖同源性,但大量微生物功能仍无法注释。基于基因上下文(如FunGeCO)或子系统(如MG-RAST)的方法通过整合基因组环境或功能网络提升注释深度。深度学习工具(如DeepFRI)结合序列与结构特征显著扩展功能信息覆盖范围。功能注释不仅可预测代谢通路、抗生素抗性基因及CRISPR-Cas系统,还能揭示氮循环、疾病相关微生物组及病毒-宿主互作等功能特征。
Ⅳ 功能谱分析
功能谱分析通过比对或非比对方法量化已知功能基因的丰度(图3中),解析微生物群落的功能动态及生态角色。比对方法(如MG-RAST、DIAMOND)基于序列相似性搜索(如BLAST、KEGG KO术语),非比对方法(如eggNOG-mapper)利用预聚类序列加速分析。DIAMOND针对短读段优化,采用种子搜索与双索引策略提升效率。
隐马尔可夫模型工具(如InterProScan、KOfamKOALA)通过预建谱模型降低计算负担,KOfamKOALA结合自适应阈值精准识别代谢功能。蛋白结构域注释(如Pfam、InterProScan)通过保守区域推断功能,支持蛋白家族分类及进化分析。
Ⅴ 通路导向分析
通路方法通过基因功能注释与分子互作解析,构建代谢通路网络(图3右)。HUMAnN通过映射读段至参考通路基因,重建宿主-环境互作的代谢模型;gutSMASH利用代谢基因簇分析跨群体功能差异。MetaCyc、KEGG、KBase等数据库提供通路注释支持,DrugBank整合药物-靶点信息,助力机制模型构建。此类方法依赖高质量参考数据库,不仅深化代谢功能理解,还凸显微生物生态系统的复杂性。
3.宏基因组的应用领域
宏基因组学的应用潜力广泛,涵盖临床病原体监测、抗生素耐药性追踪,以及生态学中的微生物功能解析、生物多样性评估和生态系统健康监测。以下分述主要应用方向。
微生物组-疾病关联研究
宏基因组数据分析已成为揭示人体微生物组组成与疾病状态关联的重要工具,可精准识别与疾病相关的微生物类群及功能,解析微生物互作动态。
-
横向研究:通过比较患者与健康人群的微生物组(如分类组成、功能谱),识别疾病相关特征。例如,2型糖尿病患者中产丁酸菌减少,硫酸盐还原与氧化应激相关功能群富集;抑郁症患者肠道菌群中氨基酸代谢通路紊乱;类风湿关节炎患者口腔与肠道微生物组中氧化还原环境及铁、硫代谢功能异常。
-
纵向研究:追踪疾病进程中微生物组的时间动态变化。例如,急性呼吸衰竭患者呼吸道与肠道菌群在治疗期间的分类与功能谱演变。
微生物多样性分析
微生物多样性分析是指特定样本的种群丰度,一般常采用香农指数(Shannon's diversity index)或phyloseq等工具。疾病当中,如低口腔/肠道菌群多样性分别与牙周炎、克罗恩病等疾病相关,但针对帕金森病、多发性硬化症等疾病的α多样性研究结果尚不一致。尽管物种组成存在个体差异,肠道微生物功能谱常呈现跨群体保守性,提示功能群组分析可能比分类单元更具临床价值。例如,基于功能互作的q2-predict-dysbiosis工具在炎症性肠病诊断中优于传统分类指标。
临床诊断应用
传统诊断方法依赖病原体培养或生物标志物检测,对复杂感染(如多重微生物感染、未知病原体)存在局限。宏基因组学通过全面解析微生物组成,为以下方向提供支持:
-
抗生素耐药性监测:通过抗性组(resistome)分析指导个性化用药,降低耐药风险。
-
病原体与毒力因子检测:如结直肠癌相关毒素基因colibactin的早期筛查。
-
疑难感染诊断:在培养阴性脓毒症、肺炎、脑膜炎等场景中展现高灵敏度与短检测周期优势。
-
菌株追踪:通过宏基因组菌株分型(如MIDAS、StrainPhlAn)指导粪菌移植治疗或医院感染暴发防控。
尽管当前临床转化面临标准化、成本与法规挑战,但测序成本下降与计算工具进步正推动其发展。
病原体传播追踪与监测
传统监测依赖分离培养,耗时耗力。宏基因组学支持无培养检测,可识别新发病原体及耐药/毒力基因,助力"一体化健康"(One Health)策略。
-
环境监测:通过水、土壤、污水等样本的系统分析,建立微生物多样性基线数据,预警新发病原体或耐药性扩散。
-
医院感染防控:结合全基因组测序追踪耐药菌株传播路径,指导隔离与精准用药。
-
空气微生物组:医院空气中机会性病原体的宏基因组监测,关联呼吸道疾病死亡率。
-
合成生物学整合:从环境样本挖掘新型酶(如脂肪酶、蛋白酶)及治疗靶点(如N-酰基酰胺合成酶与GPCR互作),推动生物工程应用。
环境健康与生态保护
-
生物修复:解析污染物降解微生物群落(如假单胞菌对烃类污染物的氧化)及关键代谢通路,指导定制化修复策略。海洋微藻(如小球藻、螺旋藻)在重金属吸附中发挥重要作用。
-
生态系统评估:监测微生物群落动态以评估气候变化、人类活动对生态的影响,支持濒危物种保护。
-
抗生素发现:从多样环境中挖掘新型抗生素基因,应对耐药菌威胁。
可持续农业
-
土壤微生物组解析:研究根际微生物组(如固氮根瘤菌、解磷菌)与作物互作,开发生物肥料,减少化肥依赖。
-
植物病害防控:鉴定抑病有益微生物,促进可持续农业实践。
-
食品安全:追踪食品生产链各环节微生物群落,优化食源性病原体检测。
-
土壤与水环境:揭示农药降解基因与微生物互作,支持污染治理;分析废水微生物助力回收技术开发。
4.公共数据库资源
-
原始数据存储库:ENA、SRA、MG-RAST等平台提供海量宏基因组数据。
-
参考基因组数据库:RefSeq、GTDB、PATRIC等涵盖不同类群,质量与更新频率各异。RefSeq定期更新且质控严格,而GenBank依赖用户提交,注释质量参差。
参考链接
- Analysis of metagenomic data. Nat Rev Methods Primers 5, 5 (2025). https://doi.org/10.1038/s43586-024-00376-6
简单分享到这里
欢迎各位佬哥佬姐关注