#!/usr/bin/env Rscript
suppressPackageStartupMessages({
library(Seurat)
library(dplyr)
library(harmony)
library(future)
library(ggplot2)
library(patchwork)
library(argparse)
library(Matrix)
library(tibble)
})
# ============================================================================
# 命令行参数解析
# ============================================================================
parser <- ArgumentParser(description = "单细胞RNA-seq分析管道 - Seurat V5 Harmony整合")
# 必需参数
parser$add_argument("--sample-file", type = "character", required = TRUE,
help = "样本信息文件路径 (制表符分隔)")
parser$add_argument("--output-dir", type = "character", default = "./results",
help = "输出目录")
# 输出参数
parser$add_argument("--output-prefix", type = "character", default = "scRNA",
help = "输出文件前缀")
# QC参数
parser$add_argument("--min-cells", type = "integer", default = 3,
help = "每个基因最少表达的细胞数")
parser$add_argument("--min-features", type = "integer", default = 200,
help = "每个细胞最少检测到的基因数")
parser$add_argument("--max-features", type = "integer", default = 6000,
help = "每个细胞最多检测到的基因数")
parser$add_argument("--max-mito", type = "double", default = 10,
help = "线粒体基因百分比阈值")
parser$add_argument("--min-counts", type = "integer", default = 500,
help = "每个细胞最少UMI数")
# 分析参数
parser$add_argument("--nfeatures", type = "integer", default = 2000,
help = "用于可变基因分析的特征数")
parser$add_argument("--npcs", type = "integer", default = 30,
help = "PCA主成分数")
parser$add_argument("--dims", type = "integer", default = 20,
help = "用于降维的PC数")
parser$add_argument("--resolution", type = "double", default = 0.5,
help = "聚类分辨率")
# Harmony参数
parser$add_argument("--harmony-vars", type = "character", default = "sample_id",
help = "Harmony整合的变量")
# 运行参数
parser$add_argument("--threads", type = "integer", default = 8,
help = "并行线程数")
parser$add_argument("--seed", type = "integer", default = 42,
help = "随机种子")
args <- parser$parse_args()
# ============================================================================
# 设置环境
# ============================================================================
cat("Seurat V5 Harmony整合管道\n")
cat("参数设置:\n")
for (arg_name in names(args)) {
cat(sprintf(" %-20s: %s\n", arg_name, args[[arg_name]]))
}
cat("\n")
# 创建输出目录
if (!dir.exists(args$output_dir)) {
dir.create(args$output_dir, recursive = TRUE)
}
# 设置并行计算
if (args$threads > 1) {
plan("multicore", workers = args$threads)
options(future.globals.maxSize = 10 * 1024^3)
}
set.seed(args$seed)
# ============================================================================
# 1. 读取样本信息
# ============================================================================
cat("步骤1: 读取样本信息\n")
sample_info <- read.delim(args$sample_file, stringsAsFactors = FALSE)
colnames(sample_info) <- c("sample_name", "sample_path", "data_format", "group", "stage")
cat("找到", nrow(sample_info), "个样本:\n")
print(sample_info)
# ============================================================================
# 2. 读取和处理单个样本
# ============================================================================
cat("步骤2: 读取样本数据\n")
read_single_sample <- function(sample_name, sample_path, data_format, group, stage) {
cat(sprintf("读取样本: %s\n", sample_name))
if (data_format == "10x") {
# 读取10x数据
data <- Read10X(data.dir = sample_path)
obj <- CreateSeuratObject(
counts = data,
project = sample_name,
min.cells = args$min_cells,
min.features = args$min_features
)
} else if (data_format == "csv") {
# 读取CSV数据
expr_matrix <- read.csv(sample_path, row.names = 1,check.names = F)
expr_matrix <- as(as.matrix(expr_matrix), "dgCMatrix")
obj <- CreateSeuratObject(
counts = expr_matrix,
project = sample_name,
min.cells = args$min_cells,
min.features = args$min_features
)
} else {
stop(sprintf("不支持的数据格式: %s", data_format))
}
# 添加元数据
obj$sample_id <- sample_name
obj$orig.ident <- sample_name
obj$group <- group
obj$stage <- stage
# 计算QC指标
obj[["percent.mt"]] <- PercentageFeatureSet(obj, pattern = "^MT-|^mt-")
obj[["percent.ribo"]] <- PercentageFeatureSet(obj, pattern = "^RP[SL]|^Rp[sl]")
# 重命名细胞
obj <- RenameCells(obj, add.cell.id = sample_name)
cat(sprintf(" 细胞数: %d, 基因数: %d\n", ncol(obj), nrow(obj)))
return(obj)
}
# 读取所有样本
seurat_list <- list()
for (i in 1:nrow(sample_info)) {
row <- sample_info[i, ]
obj <- read_single_sample(row$sample_name, row$sample_path,
row$data_format, row$group, row$stage)
seurat_list[[row$sample_name]] <- obj
}
# ============================================================================
# 3. 创建Seurat V5对象
# ============================================================================
cat("步骤3: 创建Seurat V5对象\n")
seurat_merged <- seurat_list[[1]]
if (length(seurat_list) > 1) {
for (i in 2:length(seurat_list)) {
sample_name <- names(seurat_list)[i]
cat(sprintf("合并样本 %d/%d: %s\n", i, length(seurat_list), sample_name))
seurat_merged <- merge(
x = seurat_merged,
y = seurat_list[[i]]
)
}
}
# 添加统一的元数据
metadata_df <- do.call(rbind, lapply(names(seurat_list), function(sample_name) {
obj <- seurat_list[[sample_name]]
# 直接从元数据中获取(如果存在)
if ("group" %in% colnames(obj@meta.data) && "stage" %in% colnames(obj@meta.data)) {
data.frame(
row.names = colnames(obj),
sample_id = sample_name,
group = obj$group,
stage = obj$stage,
stringsAsFactors = FALSE
)
} else {
# 如果元数据中没有,使用默认值
n_cells <- ncol(obj)
data.frame(
row.names = colnames(obj),
sample_id = rep(sample_name, n_cells),
group = rep("Unknown", n_cells),
stage = rep("Unknown", n_cells),
stringsAsFactors = FALSE
)
}
}))
seurat_merged <- AddMetaData(seurat_merged, metadata = metadata_df)
cat("合并后总细胞数:", ncol(seurat_merged), "\n")
cat("合并后基因数:", nrow(seurat_merged), "\n")
# ============================================================================
# 4. 质量控制
# ============================================================================
cat("步骤4: 质量控制\n")
# 重新计算QC指标(为了确保一致)
seurat_merged[["percent.mt"]] <- PercentageFeatureSet(seurat_merged, pattern = "^MT-|^mt-")
original_cells <- ncol(seurat_merged)
# 过滤细胞
seurat_filtered <- subset(seurat_merged,
subset = nFeature_RNA > args$min_features &
nFeature_RNA < args$max_features &
nCount_RNA > args$min_counts &
percent.mt < args$max_mito)
cat("过滤前:", original_cells, "个细胞\n")
cat("过滤后:", ncol(seurat_filtered), "个细胞\n")
cat("过滤掉:", original_cells - ncol(seurat_filtered), "个细胞\n")
# QC可视化
qc_plots <- VlnPlot(seurat_filtered,
features = c("nFeature_RNA", "nCount_RNA", "percent.mt"),
group.by = "sample_id",
pt.size = 0.1,
ncol = 3)
ggsave(file.path(args$output_dir, paste0(args$output_prefix, "_QC_violin.pdf")),
qc_plots, width = 15, height = 5)
# ============================================================================
# 5. 数据标准化和预处理
# ============================================================================
cat("步骤5: 数据标准化和预处理\n")
# 标准化
seurat_filtered <- NormalizeData(seurat_filtered)
# 寻找高变基因
seurat_filtered <- FindVariableFeatures(seurat_filtered,
selection.method = "vst",
nfeatures = args$nfeatures)
# 缩放数据
seurat_filtered <- ScaleData(seurat_filtered)
cat("找到", length(VariableFeatures(seurat_filtered)), "个高变基因\n")
# 高变基因可视化
var_plot <- VariableFeaturePlot(seurat_filtered)
top10 <- head(VariableFeatures(seurat_filtered), 10)
var_plot <- LabelPoints(plot = var_plot, points = top10, repel = TRUE)
ggsave(file.path(args$output_dir, paste0(args$output_prefix, "_variable_genes.pdf")),
var_plot, width = 10, height = 8)
# ============================================================================
# 6. PCA降维
# ============================================================================
cat("步骤6: PCA降维\n")
# 运行PCA
seurat_filtered <- RunPCA(seurat_filtered,
features = VariableFeatures(object = seurat_filtered))
# 肘部图
elbow_plot <- ElbowPlot(seurat_filtered, ndims = 50)
ggsave(file.path(args$output_dir, paste0(args$output_prefix, "_elbow_plot.pdf")),
elbow_plot, width = 8, height = 6)
cat("PCA计算完成\n")
# ============================================================================
# 7. Seurat V5 Harmony整合
# ============================================================================
cat("步骤7: Seurat V5 Harmony整合\n")
cat("使用 IntegrateLayers 进行 Harmony 整合...\n")
# 关键步骤:为每个样本设置layer
seurat_filtered$orig.ident <- seurat_filtered$sample_id
# 执行Harmony整合
seurat_harmony <- IntegrateLayers(
object = seurat_filtered,
method = HarmonyIntegration,
orig.reduction = "pca",
new.reduction = "harmony",
verbose = FALSE,
assay = "RNA"
)
cat("Harmony整合完成\n")
# 检查整合结果
cat("可用降维方法:", names(seurat_harmony@reductions), "\n")
# ============================================================================
# 8. UMAP降维
# ============================================================================
cat("步骤8: UMAP降维\n")
# 基于Harmony整合后的空间运行UMAP
seurat_harmony <- RunUMAP(seurat_harmony,
reduction = "harmony",
dims = 1:min(args$dims, ncol(seurat_harmony[["harmony"]])),
reduction.name = "umap.harmony",
reduction.key = "UMAPHARMONY_")
# 可视化
umap_by_sample <- DimPlot(seurat_harmony,
reduction = "umap.harmony",
group.by = "sample_id",
pt.size = 0.5,
label = FALSE)
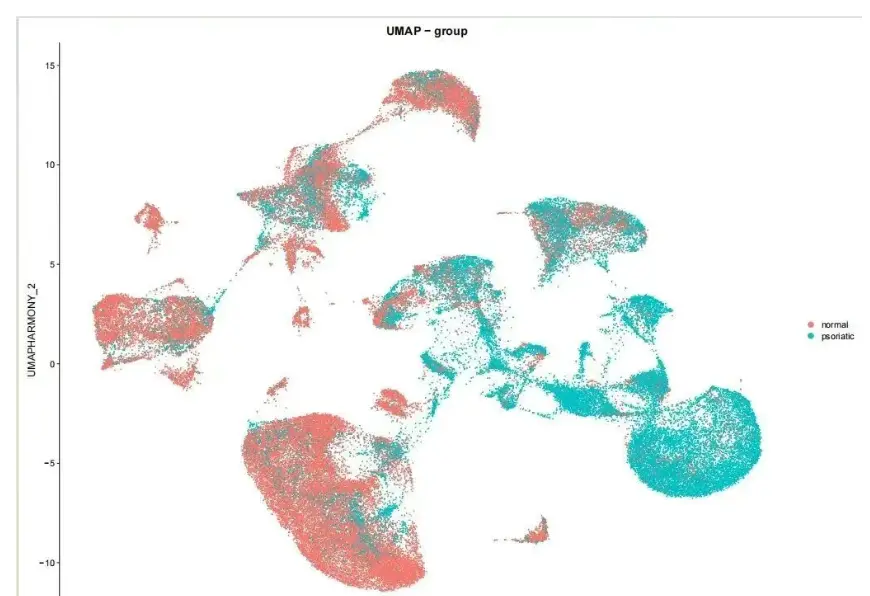
umap_by_group <- DimPlot(seurat_harmony,
reduction = "umap.harmony",
group.by = "group",
pt.size = 0.5,
label = FALSE)
umap_by_stage <- DimPlot(seurat_harmony,
reduction = "umap.harmony",
group.by = "stage",
pt.size = 0.5,
label = FALSE)
# 组合图
combined_umap <- umap_by_sample + umap_by_group + umap_by_stage
ggsave(file.path(args$output_dir, paste0(args$output_prefix, "_UMAP_harmony.pdf")),
combined_umap, width = 15, height = 5)
# 按样本分面显示
faceted_umap <- DimPlot(seurat_harmony,
reduction = "umap.harmony",
group.by = "sample_id",
split.by = "sample_id",
ncol = min(4, length(unique(seurat_harmony$sample_id))),
pt.size = 0.5)
ggsave(file.path(args$output_dir, paste0(args$output_prefix, "_UMAP_by_sample.pdf")),
faceted_umap,
width = min(15, 4 * length(unique(seurat_harmony$sample_id))),
height = 10)
# ============================================================================
# 9. 细胞聚类
# ============================================================================
cat("步骤9: 细胞聚类\n")
# 基于Harmony整合后的空间构建邻居图
seurat_harmony <- FindNeighbors(seurat_harmony,
reduction = "harmony",
dims = 1:min(args$dims, ncol(seurat_harmony[["harmony"]])))
# 聚类分析
seurat_harmony <- FindClusters(seurat_harmony, resolution = args$resolution)
# 聚类可视化
cluster_plot <- DimPlot(seurat_harmony,
reduction = "umap.harmony",
group.by = "seurat_clusters",
label = TRUE,
pt.size = 0.5) +
ggtitle(paste("Clusters (resolution =", args$resolution, ")"))
ggsave(file.path(args$output_dir, paste0(args$output_prefix, "_clusters.pdf")),
cluster_plot, width = 10, height = 8)
cat("找到", length(unique(seurat_harmony$seurat_clusters)), "个cluster\n")
# 聚类统计
cluster_stats <- as.data.frame(table(seurat_harmony$seurat_clusters))
colnames(cluster_stats) <- c("Cluster", "Cell_Count")
cluster_stats$Percentage <- round(cluster_stats$Cell_Count / sum(cluster_stats$Cell_Count) * 100, 2)
write.csv(cluster_stats,
file.path(args$output_dir, paste0(args$output_prefix, "_cluster_stats.csv")),
row.names = FALSE)
cat("聚类统计:\n")
print(cluster_stats)
# ============================================================================
# 10. 差异表达分析
# ============================================================================
cat("步骤10: 差异表达分析\n")
seurat_harmony = JoinLayers(seurat_harmony)
# 设置默认ident为cluster
Idents(seurat_harmony) <- "seurat_clusters"
# 寻找所有cluster的marker基因
cat("寻找marker基因...\n")
all_markers <- FindAllMarkers(seurat_harmony,
only.pos = TRUE,
min.pct = 0.25,
logfc.threshold = 0.25,
test.use = "wilcox")
# 保存所有marker基因
write.csv(all_markers,
file.path(args$output_dir, paste0(args$output_prefix, "_all_markers.csv")),
row.names = FALSE)
cat("找到", nrow(all_markers), "个marker基因\n")
# 提取每个cluster的前10个marker基因
top10_markers <- all_markers %>%
group_by(cluster) %>%
top_n(n = 10, wt = avg_log2FC)
write.csv(top10_markers,
file.path(args$output_dir, paste0(args$output_prefix, "_top10_markers.csv")),
row.names = FALSE)
# 热图可视化(显示每个cluster的前5个marker基因)
top5_markers <- all_markers %>%
group_by(cluster) %>%
top_n(n = 5, wt = avg_log2FC)
# 创建热图
if (nrow(top5_markers) > 0) {
heatmap_genes <- unique(top5_markers$gene)
if (length(heatmap_genes) > 0) {
heatmap_plot <- DoHeatmap(seurat_harmony,
features = heatmap_genes,
group.by = "seurat_clusters",
size = 3) +
theme(axis.text.y = element_text(size = 6))
ggsave(file.path(args$output_dir, paste0(args$output_prefix, "_marker_heatmap.pdf")),
heatmap_plot,
width = 12,
height = max(8, length(heatmap_genes) * 0.15))
}
}
# ============================================================================
# 11. 保存结果
# ============================================================================
cat("步骤11: 保存结果\n")
# 保存RDS文件
rds_file <- file.path(args$output_dir, paste0(args$output_prefix, "_harmony_integrated.rds"))
saveRDS(seurat_harmony, file = rds_file)
cat("保存RDS文件:", rds_file, "\n")
# 保存元数据
metadata <- seurat_harmony@meta.data
write.csv(metadata,
file.path(args$output_dir, paste0(args$output_prefix, "_metadata.csv")),
row.names = TRUE)
# 保存整合信息
integration_summary <- data.frame(
Parameter = c("Samples", "Cells_Before_QC", "Cells_After_QC",
"Genes", "Clusters", "Resolution", "Integration_Method"),
Value = c(nrow(sample_info),
original_cells,
ncol(seurat_harmony),
nrow(seurat_harmony),
length(unique(seurat_harmony$seurat_clusters)),
args$resolution,
"Harmony (IntegrateLayers)")
)
write.csv(integration_summary,
file.path(args$output_dir, paste0(args$output_prefix, "_integration_summary.csv")),
row.names = FALSE)
#!/usr/bin/env python3
# -*- coding: utf-8 -*-
###zhaoyunfei
###20251227
"""
单细胞RNA-seq分析管道 - Scanpy + Harmony
功能:多样本整合、Harmony批次校正、差异基因分析
输入:sample.txt文件
格式:样本名称\t样本路径\t数据格式\tgroup\tstage
"""
import argparse
import os
import sys
import pandas as pd
import numpy as np
import scanpy as sc
import scanpy.external as sce
import harmonypy as hm
import anndata as ad
import matplotlib.pyplot as plt
import matplotlib
import seaborn as sns
from scipy import sparse, stats
import warnings
import logging
from typing import List, Dict, Tuple, Optional
# 设置日志
logging.basicConfig(
level=logging.INFO,
format='%(asctime)s - %(levelname)s - %(message)s',
datefmt='%Y-%m-%d %H:%M:%S'
)
logger = logging.getLogger(__name__)
# 设置随机种子
np.random.seed(42)
# 设置matplotlib
plt.rcParams['pdf.fonttype'] = 42
plt.rcParams['ps.fonttype'] = 42
plt.rcParams['font.sans-serif'] = ['Arial', 'DejaVu Sans']
plt.rcParams['axes.unicode_minus'] = False
plt.rcParams['figure.dpi'] = 100
# 设置scanpy
sc.settings.verbosity = 3
sc.settings.set_figure_params(
dpi_save=300,
facecolor='white',
frameon=False,
transparent=False
)
# ============================================================================
# 命令行参数解析
# ============================================================================
def parse_arguments():
parser = argparse.ArgumentParser(
description='单细胞RNA-seq分析管道 (Scanpy + Harmony)',
formatter_class=argparse.RawDescriptionHelpFormatter,
epilog="""
示例:
python scRNA_harmony.py --sample-file sample.txt --output-dir ./results
python scRNA_harmony.py --sample-file sample.txt --harmony-key sample_id --resolution 0.6
"""
)
# 必需参数
parser.add_argument('--sample-file', type=str, required=True,
help='样本信息文件路径 (制表符分隔)')
# 输出参数
parser.add_argument('--output-dir', type=str, default='./scanpy_harmony_results',
help='输出目录 (默认: ./scanpy_harmony_results)')
parser.add_argument('--output-prefix', type=str, default='scRNA',
help='输出文件前缀 (默认: scRNA)')
# 质量控制参数
parser.add_argument('--min-cells', type=int, default=3,
help='每个基因最少表达的细胞数 (默认: 3)')
parser.add_argument('--min-genes', type=int, default=200,
help='每个细胞最少检测到的基因数 (默认: 200)')
parser.add_argument('--max-genes', type=int, default=7000,
help='每个细胞最多检测到的基因数 (默认: 7000)')
parser.add_argument('--max-mito', type=float, default=10.0,
help='线粒体基因百分比阈值 (默认: 10%%)')
parser.add_argument('--min-counts', type=int, default=500,
help='每个细胞最少UMI数 (默认: 500)')
# 分析参数
parser.add_argument('--n-top-genes', type=int, default=2000,
help='用于可变基因分析的特征数 (默认: 2000)')
parser.add_argument('--n-pcs', type=int, default=50,
help='PCA主成分数 (默认: 50)')
parser.add_argument('--n-neighbors', type=int, default=15,
help='KNN邻居数 (默认: 15)')
parser.add_argument('--resolution', type=float, default=0.5,
help='聚类分辨率 (默认: 0.5)')
# Harmony参数
parser.add_argument('--harmony-key', type=str, default='sample_id',
help='Harmony批次校正的关键字段 (默认: sample_id)')
parser.add_argument('--harmony-vars', type=str, nargs='+', default=None,
help='Harmony整合的多个变量')
# 运行参数
parser.add_argument('--threads', type=int, default=8,
help='并行线程数 (默认: 4)')
parser.add_argument('--seed', type=int, default=42,
help='随机种子 (默认: 42)')
# 过滤参数
parser.add_argument('--skip-doublet', action='store_true',
help='跳过双细胞检测')
parser.add_argument('--skip-celltype', action='store_true',
help='跳过细胞类型注释')
return parser.parse_args()
# ============================================================================
# 数据读取函数
# ============================================================================
def read_sample_data(sample_info: pd.Series) -> Optional[ad.AnnData]:
"""
读取单个样本数据
"""
sample_name = sample_info['sample_name']
sample_path = sample_info['sample_path']
data_format = sample_info['data_format']
logger.info(f"读取样本: {sample_name}")
try:
if data_format == '10x':
# 读取10x数据
if not os.path.exists(sample_path):
logger.error(f"10x目录不存在: {sample_path}")
return None
# 查找10x文件
matrix_file = None
for f in ['matrix.mtx.gz', 'matrix.mtx']:
potential_path = os.path.join(sample_path, f)
if os.path.exists(potential_path):
matrix_file = potential_path
break
if matrix_file is None:
logger.error(f"未找到10x矩阵文件: {sample_path}")
return None
# 读取数据
adata = sc.read_10x_mtx(
sample_path,
var_names='gene_symbols',
cache=False
)
elif data_format == 'csv':
# 读取CSV数据
if not os.path.exists(sample_path):
logger.error(f"CSV文件不存在: {sample_path}")
return None
# 读取表达矩阵
expr_matrix = pd.read_csv(sample_path, index_col=0)
# 转换为稀疏矩阵
if isinstance(expr_matrix, pd.DataFrame):
X = sparse.csr_matrix(expr_matrix.values.T)
else:
X = expr_matrix
# 创建AnnData对象
adata = ad.AnnData(
X=X,
var=pd.DataFrame(index=expr_matrix.index),
obs=pd.DataFrame(index=expr_matrix.columns)
)
elif data_format == 'h5ad':
# 读取h5ad文件
if not os.path.exists(sample_path):
logger.error(f"h5ad文件不存在: {sample_path}")
return None
adata = sc.read_h5ad(sample_path)
else:
logger.error(f"不支持的数据格式: {data_format}")
return None
# 添加样本信息
adata.obs['sample_id'] = sample_info['sample_name']
adata.obs['group'] = sample_info['group']
adata.obs['stage'] = sample_info['stage']
adata.obs['dataset'] = f"{sample_info['group']}_{sample_info['stage']}"
# 重命名细胞barcodes
adata.obs_names = [f"{sample_info['sample_name']}_{barcode}"
for barcode in adata.obs_names]
logger.info(f" ✓ 细胞数: {adata.n_obs:,}, 基因数: {adata.n_vars:,}")
return adata
except Exception as e:
logger.error(f"读取样本 {sample_name} 失败: {str(e)}")
return None
# ============================================================================
# 质量控制函数
# ============================================================================
def quality_control(adata: ad.AnnData, args) -> Tuple[ad.AnnData, Dict]:
"""
执行质量控制
"""
logger.info("执行质量控制...")
# 计算QC指标
adata.var['mt'] = adata.var_names.str.startswith(('MT-', 'mt-'))
adata.var['ribo'] = adata.var_names.str.startswith(('RPS', 'RPL', 'Rp', 'rp'))
sc.pp.calculate_qc_metrics(
adata,
qc_vars=['mt', 'ribo'],
percent_top=None,
log1p=False,
inplace=True
)
# 保存原始统计
original_stats = {
'n_cells': adata.n_obs,
'n_genes': adata.n_vars,
'total_counts': adata.obs['total_counts'].sum()
}
# 可视化QC指标
logger.info("生成QC可视化...")
# 创建QC图
fig, axes = plt.subplots(1, 3, figsize=(15, 4))
metrics = [
('n_genes_by_counts', 'Number of genes', args.min_genes, args.max_genes),
('total_counts', 'Total counts', args.min_counts, None),
('pct_counts_mt', 'Mitochondrial %', None, args.max_mito)
]
for i, (metric, title, min_val, max_val) in enumerate(metrics):
axes[i].hist(adata.obs[metric], bins=50, edgecolor='black', alpha=0.7)
if min_val:
axes[i].axvline(x=min_val, color='red', linestyle='--', linewidth=1.5)
if max_val:
axes[i].axvline(x=max_val, color='red', linestyle='--', linewidth=1.5)
axes[i].set_xlabel(title)
axes[i].set_ylabel('Number of cells')
axes[i].set_title(f'{title} distribution')
axes[i].grid(True, alpha=0.3)
plt.tight_layout()
plt.savefig(os.path.join(args.output_dir, f'{args.output_prefix}_QC_distribution.pdf'),
bbox_inches='tight', dpi=300)
plt.close()
# 应用过滤
logger.info(f"过滤前: {adata.n_obs:,} 个细胞")
# 基因过滤
sc.pp.filter_cells(adata, min_genes=args.min_genes)
sc.pp.filter_cells(adata, max_genes=args.max_genes)
# UMI过滤
if 'total_counts' in adata.obs.columns:
adata = adata[adata.obs['total_counts'] > args.min_counts, :]
# 线粒体基因过滤
if 'pct_counts_mt' in adata.obs.columns:
adata = adata[adata.obs['pct_counts_mt'] < args.max_mito, :]
# 基因过滤
sc.pp.filter_genes(adata, min_cells=args.min_cells)
logger.info(f"过滤后: {adata.n_obs:,} 个细胞")
logger.info(f"过滤掉: {original_stats['n_cells'] - adata.n_obs:,} 个细胞")
logger.info(f"剩余基因: {adata.n_vars:,}")
# 更新统计信息
qc_stats = {
**original_stats,
'n_cells_after_qc': adata.n_obs,
'n_genes_after_qc': adata.n_vars,
'cells_removed': original_stats['n_cells'] - adata.n_obs,
'genes_removed': original_stats['n_genes'] - adata.n_vars
}
return adata, qc_stats
# ============================================================================
# 数据预处理函数
# ============================================================================
def preprocess_data(adata: ad.AnnData, args) -> ad.AnnData:
"""
数据标准化和预处理
"""
logger.info("数据标准化和预处理...")
# 标准化
sc.pp.normalize_total(adata, target_sum=1e4)
sc.pp.log1p(adata)
# 高变基因选择
logger.info(f"选择 {args.n_top_genes} 个高变基因...")
sc.pp.highly_variable_genes(
adata,
n_top_genes=args.n_top_genes,
flavor='seurat',
batch_key=args.harmony_key if args.harmony_key in adata.obs.columns else None
)
# 缩放数据
logger.info("缩放数据...")
sc.pp.scale(adata, max_value=10)
# 可视化高变基因
sc.pl.highly_variable_genes(adata, show=False)
plt.savefig(os.path.join(args.output_dir, f'{args.output_prefix}_highly_variable_genes.pdf'),
bbox_inches='tight', dpi=300)
return adata
# ============================================================================
# Harmony整合函数
# ============================================================================
def run_harmony_integration(adata: ad.AnnData, args) -> ad.AnnData:
"""
执行Harmony批次校正
"""
logger.info("执行Harmony批次校正...")
# 检查批次变量
if args.harmony_key not in adata.obs.columns:
logger.warning(f"批次变量 '{args.harmony_key}' 不存在,跳过Harmony整合")
return adata
n_batches = len(adata.obs[args.harmony_key].unique())
if n_batches <= 1:
logger.warning("只有一个批次,跳过Harmony整合")
return adata
logger.info(f"批次数量: {n_batches}")
# 运行PCA
logger.info(f"运行PCA ({args.n_pcs}个主成分)...")
sc.tl.pca(adata, svd_solver='arpack', n_comps=args.n_pcs, random_state=args.seed)
# 运行Harmony整合
logger.info(f"使用sce.pp.harmony_integrate进行整合...")
try:
# 使用scanpy.external的harmony_integrate
sce.pp.harmony_integrate(
adata,
key=args.harmony_key,
basis='X_pca',
adjusted_basis='X_pca_harmony',
max_iter_harmony=50,
random_state=args.seed
)
# 检查是否成功
if 'X_pca_harmony' in adata.obsm:
logger.info("✓ Harmony整合成功")
# 将harmony结果设为默认的PCA
adata.obsm['X_pca'] = adata.obsm['X_pca_harmony']
else:
logger.warning("Harmony整合可能失败,使用原始PCA")
except Exception as e:
logger.error(f"Harmony整合失败: {str(e)}")
logger.info("使用原始PCA继续分析")
return adata
# ============================================================================
# 降维和聚类函数
# ============================================================================
def run_dim_reduction_and_clustering(adata: ad.AnnData, args) -> ad.AnnData:
"""
执行降维和聚类分析
"""
logger.info("执行降维和聚类分析...")
# 检查是否有PCA结果
if 'X_pca' not in adata.obsm:
logger.info("未找到PCA结果,重新计算PCA...")
sc.tl.pca(adata, svd_solver='arpack', n_comps=args.n_pcs, random_state=args.seed)
# 肘部图
logger.info("生成肘部图...")
sc.pl.pca_variance_ratio(adata, log=True, n_pcs=min(50, adata.n_obs-1), show=False)
plt.savefig(os.path.join(args.output_dir, f'{args.output_prefix}_pca_variance.pdf'),
bbox_inches='tight', dpi=300)
# 计算邻居图
logger.info(f"计算邻居图 (n_neighbors={args.n_neighbors})...")
sc.pp.neighbors(
adata,
n_pcs=min(args.n_pcs, adata.obsm['X_pca'].shape[1]),
n_neighbors=args.n_neighbors,
random_state=args.seed
)
# UMAP降维
logger.info("计算UMAP...")
sc.tl.umap(adata, random_state=args.seed)
# 聚类
logger.info(f"Leiden聚类 (resolution={args.resolution})...")
sc.tl.leiden(
adata,
resolution=args.resolution,
random_state=args.seed,
key_added='cluster'
)
# 可视化
logger.info("生成可视化图...")
# UMAP图
# 按不同分组着色
color_bys = ['sample_id', 'group', 'stage', 'cluster', 'dataset']
for i, color_by in enumerate(color_bys[:5]):
if color_by in adata.obs.columns:
sc.pl.umap(
adata,
color=color_by,
size=20,
show=False,
legend_loc='on data' if len(adata.obs[color_by].unique()) <= 10 else 'right margin'
)
# 第六个图显示批次效应
if args.harmony_key in adata.obs.columns and len(adata.obs[args.harmony_key].unique()) > 1:
sc.pl.umap(
adata,
color=args.harmony_key,
size=20,
show=False,
legend_loc='right margin'
)
plt.savefig(os.path.join(args.output_dir, f'{args.output_prefix}_umap_overview.pdf'),
bbox_inches='tight', dpi=300)
# 单独保存聚类图
sc.pl.umap(
adata,
color='cluster',
size=30,
legend_loc='on data',
title=f'Clusters (resolution={args.resolution})',
show=False,
)
plt.savefig(os.path.join(args.output_dir, f'{args.output_prefix}_umap_clusters.pdf'),
bbox_inches='tight', dpi=300)
return adata
# ============================================================================
# 差异表达分析函数
# ============================================================================
def run_differential_expression(adata: ad.AnnData, args) -> pd.DataFrame:
"""
执行差异表达分析
"""
logger.info("执行差异表达分析...")
# 检查是否有聚类结果
if 'cluster' not in adata.obs.columns:
logger.warning("未找到聚类结果,跳过差异表达分析")
return pd.DataFrame()
# 寻找marker基因
logger.info("寻找每个cluster的marker基因...")
sc.tl.rank_genes_groups(
adata,
'cluster',
method='wilcoxon',
use_raw=False,
pts=True
)
# 提取所有marker基因
all_markers = pd.DataFrame()
clusters = sorted(adata.obs['cluster'].unique())
for cluster in clusters:
try:
cluster_markers = sc.get.rank_genes_groups_df(adata, group=cluster)
if not cluster_markers.empty:
cluster_markers['cluster'] = cluster
# 过滤显著marker基因
cluster_markers = cluster_markers[
(cluster_markers['pvals_adj'] < 0.05) &
(cluster_markers['logfoldchanges'] > 0.25) &
(cluster_markers['pct_nz_group'] > 0.1)
]
all_markers = pd.concat([all_markers, cluster_markers], ignore_index=True)
except Exception as e:
logger.warning(f"处理cluster {cluster}时出错: {str(e)}")
if all_markers.empty:
logger.warning("未找到显著的marker基因")
return pd.DataFrame()
# 排序和保存
all_markers = all_markers.sort_values(['cluster', 'logfoldchanges'],
ascending=[True, False])
# 保存所有marker基因
output_file = os.path.join(args.output_dir, f'{args.output_prefix}_all_markers.csv')
all_markers.to_csv(output_file, index=False)
logger.info(f"保存所有marker基因到: {output_file}")
logger.info(f"找到 {len(all_markers)} 个marker基因")
# 保存每个cluster的前10个marker基因
top10_markers = all_markers.groupby('cluster').head(10)
top10_file = os.path.join(args.output_dir, f'{args.output_prefix}_top10_markers.csv')
top10_markers.to_csv(top10_file, index=False)
# 生成热图
logger.info("生成marker基因热图...")
try:
# 选择每个cluster的前5个marker基因
top5_genes = []
for cluster in clusters:
cluster_genes = all_markers[all_markers['cluster'] == cluster].head(5)['names'].tolist()
top5_genes.extend(cluster_genes[:5])
top5_genes = list(dict.fromkeys(top5_genes)) # 去重
if len(top5_genes) > 0:
# 热图
sc.pl.heatmap(
adata,
var_names=top5_genes[:50], # 最多50个基因
groupby='cluster',
swap_axes=True,
figsize=(12, max(8, len(top5_genes) * 0.3)),
cmap='RdBu_r',
dendrogram=True,
show=False,
save=f'{args.output_prefix}_marker_heatmap.pdf'
)
# 点图
sc.pl.dotplot(
adata,
var_names=top5_genes[:30], # 最多30个基因
groupby='cluster',
standard_scale='var',
figsize=(10, 8),
show=False,
save=f'{args.output_prefix}_marker_dotplot.pdf'
)
except Exception as e:
logger.error(f"生成热图时出错: {str(e)}")
return all_markers
# ============================================================================
# 保存结果函数
# ============================================================================
def save_results(adata: ad.AnnData, sample_info: pd.DataFrame,
qc_stats: Dict, all_markers: pd.DataFrame, args):
"""
保存所有分析结果
"""
logger.info("保存分析结果...")
# 保存AnnData对象
h5ad_file = os.path.join(args.output_dir, f'{args.output_prefix}_processed.h5ad')
adata.write(h5ad_file)
logger.info(f"保存AnnData对象到: {h5ad_file}")
# 保存元数据
metadata_file = os.path.join(args.output_dir, f'{args.output_prefix}_metadata.csv')
adata.obs.to_csv(metadata_file)
logger.info(f"保存元数据到: {metadata_file}")
# 保存聚类统计
if 'cluster' in adata.obs.columns:
cluster_stats = adata.obs['cluster'].value_counts().reset_index()
cluster_stats.columns = ['Cluster', 'Cell_Count']
cluster_stats['Percentage'] = (cluster_stats['Cell_Count'] /
cluster_stats['Cell_Count'].sum() * 100).round(2)
cluster_stats = cluster_stats.sort_values('Cluster')
cluster_file = os.path.join(args.output_dir, f'{args.output_prefix}_cluster_stats.csv')
cluster_stats.to_csv(cluster_file, index=False)
logger.info(f"保存聚类统计到: {cluster_file}")
logger.info("聚类统计:")
logger.info(f"\n{cluster_stats.to_string(index=False)}")
# 保存分析总结
summary_data = {
'Parameter': [
'Samples', 'Cells_Before_QC', 'Cells_After_QC',
'Genes_After_QC', 'Clusters', 'Resolution',
'Harmony_Key', 'Marker_Genes_Found'
],
'Value': [
len(sample_info),
qc_stats.get('n_cells', 'N/A'),
qc_stats.get('n_cells_after_qc', 'N/A'),
qc_stats.get('n_genes_after_qc', 'N/A'),
len(adata.obs['cluster'].unique()) if 'cluster' in adata.obs.columns else 'N/A',
args.resolution,
args.harmony_key,
len(all_markers) if not all_markers.empty else 0
]
}
summary_df = pd.DataFrame(summary_data)
summary_file = os.path.join(args.output_dir, f'{args.output_prefix}_analysis_summary.csv')
summary_df.to_csv(summary_file, index=False)
logger.info(f"保存分析总结到: {summary_file}")
# 生成文本报告
report_file = os.path.join(args.output_dir, f'{args.output_prefix}_analysis_report.txt')
with open(report_file, 'w') as f:
f.write("=" * 60 + "\n")
f.write("单细胞RNA-seq分析报告 (Scanpy + Harmony)\n")
f.write("=" * 60 + "\n\n")
f.write("分析时间: " + pd.Timestamp.now().strftime("%Y-%m-%d %H:%M:%S") + "\n")
f.write(f"Scanpy版本: {sc.__version__}\n")
f.write(f"Harmony版本: {hm.__version__}\n\n")
f.write("1. 样本信息\n")
f.write("-" * 40 + "\n")
f.write(sample_info.to_string() + "\n\n")
f.write("2. 质量控制\n")
f.write("-" * 40 + "\n")
f.write(f"原始细胞数: {qc_stats.get('n_cells', 'N/A'):,}\n")
f.write(f"质控后细胞数: {qc_stats.get('n_cells_after_qc', 'N/A'):,}\n")
f.write(f"过滤细胞数: {qc_stats.get('cells_removed', 'N/A'):,}\n")
f.write(f"质控后基因数: {qc_stats.get('n_genes_after_qc', 'N/A'):,}\n\n")
f.write("3. 分析参数\n")
f.write("-" * 40 + "\n")
for key, value in vars(args).items():
f.write(f"{key:20}: {value}\n")
logger.info(f"保存分析报告到: {report_file}")
# ============================================================================
# 主函数
# ============================================================================
def main():
# 解析参数
args = parse_arguments()
print("\n" + "=" * 60)
print("单细胞RNA-seq分析管道 (Scanpy + Harmony)")
print("=" * 60 + "\n")
print("参数设置:")
for key, value in vars(args).items():
print(f" {key:20}: {value}")
print()
# 创建输出目录
os.makedirs(args.output_dir, exist_ok=True)
print(f"输出目录: {args.output_dir}")
# 设置随机种子
np.random.seed(args.seed)
# ========================================================================
# 1. 读取样本信息
# ========================================================================
print("\n" + "=" * 60)
print("步骤1: 读取样本信息")
print("=" * 60)
if not os.path.exists(args.sample_file):
print(f"错误: 样本文件不存在 - {args.sample_file}")
sys.exit(1)
try:
sample_info = pd.read_csv(args.sample_file, sep='\t', header=None)
sample_info.columns = ['sample_name', 'sample_path', 'data_format', 'group', 'stage']
print(f"成功读取 {len(sample_info)} 个样本:")
print(sample_info.to_string(index=False))
except Exception as e:
print(f"读取样本信息失败: {str(e)}")
sys.exit(1)
# ========================================================================
# 2. 读取所有样本数据
# ========================================================================
print("\n" + "=" * 60)
print("步骤2: 读取样本数据")
print("=" * 60)
adatas = []
for idx, row in sample_info.iterrows():
adata = read_sample_data(row)
if adata is not None:
adatas.append(adata)
if len(adatas) == 0:
print("错误: 没有成功读取任何样本数据")
sys.exit(1)
print(f"\n成功读取 {len(adatas)}/{len(sample_info)} 个样本")
# ========================================================================
# 3. 合并数据
# ========================================================================
print("\n合并样本数据...")
if len(adatas) == 1:
adata_combined = adatas[0]
else:
adata_combined = ad.concat(adatas, join='outer', label='batch_id')
adata_combined.var_names_make_unique()
print(f"合并后总细胞数: {adata_combined.n_obs:,}")
print(f"合并后基因数: {adata_combined.n_vars:,}")
# ========================================================================
# 4. 质量控制
# ========================================================================
print("\n" + "=" * 60)
print("步骤3: 质量控制")
print("=" * 60)
adata_filtered, qc_stats = quality_control(adata_combined, args)
# ========================================================================
# 5. 数据预处理
# ========================================================================
print("\n" + "=" * 60)
print("步骤4: 数据预处理")
print("=" * 60)
adata_processed = preprocess_data(adata_filtered, args)
# ========================================================================
# 6. Harmony批次校正
# ========================================================================
print("\n" + "=" * 60)
print("步骤5: Harmony批次校正")
print("=" * 60)
adata_harmony = run_harmony_integration(adata_processed, args)
# ========================================================================
# 7. 降维和聚类
# ========================================================================
print("\n" + "=" * 60)
print("步骤6: 降维和聚类")
print("=" * 60)
adata_clustered = run_dim_reduction_and_clustering(adata_harmony, args)
# ========================================================================
# 8. 差异表达分析
# ========================================================================
print("\n" + "=" * 60)
print("步骤7: 差异表达分析")
print("=" * 60)
all_markers = run_differential_expression(adata_clustered, args)
# ========================================================================
# 9. 保存结果
# ========================================================================
print("\n" + "=" * 60)
print("步骤8: 保存结果")
print("=" * 60)
save_results(adata_clustered, sample_info, qc_stats, all_markers, args)
# ========================================================================
# 完成
# ========================================================================
print("\n" + "=" * 60)
print("分析完成!")
print("=" * 60 + "\n")
print("输出文件:")
print(f" 1. H5AD文件: {args.output_prefix}_processed.h5ad")
print(f" 2. 元数据: {args.output_prefix}_metadata.csv")
if not all_markers.empty:
print(f" 3. Marker基因: {args.output_prefix}_all_markers.csv")
print(f" 4. Top10 Marker: {args.output_prefix}_top10_markers.csv")
print(f" 5. 聚类统计: {args.output_prefix}_cluster_stats.csv")
print(f" 6. 分析总结: {args.output_prefix}_analysis_summary.csv")
print(f" 7. 分析报告: {args.output_prefix}_analysis_report.txt")
print(f" 8. 可视化图: {args.output_prefix}_*.pdf")
print("\n使用以下命令重新加载数据:")
print(f" adata = sc.read_h5ad('{os.path.join(args.output_dir, args.output_prefix + '_processed.h5ad')}')")
# ============================================================================
# 运行主函数
# ============================================================================
if __name__ == "__main__":
main()