概述
论文地址:https://arxiv.org/abs/2309.16235
虽然近年来技术创新和变革日新月异,从根本上改变了我们对生物化学过程的认识,但化学领域仍花费大量时间和金钱--"10 年 "和 "3000 亿"--将新产品推向市场。这是由于实验室实验的高失败率、化学探索的广阔空间以及包括意外发现在内的强大运气成分。常见的情况是,设计一种分子,设计一条合成路线,并根据各种理论花费大量时间进行合成,却发现无法实现预期功能。然后,经过反复实验,可以说是机缘巧合的发现促成了新产品的诞生,如药品。
在此背景下,出现了大规模语言模型,也出现了能够像人类一样理解和生成文本的技术,并在多个领域取得了成功。化学领域也有这方面的潜力。分子也可以用语言来表示,这有可能加速分子设计和发现过程。近年来,大规模语言模型在处理从蛋白质折叠到小分子、多肽和聚合物设计等化学语言方面取得了卓越的成果。

但大规模语言模型究竟是什么呢?简单地说,它们是机器学习模型,能够理解文本片段并据此做出连续推断。这些模型通过学习单词序列的概率分布来完成文本生成和语言翻译等任务。
为什么语言模型对化学有用?通过学习表示化学结构,这些模型有助于探索化学空间,并设计出具有特定功能特性的分子。此外,弥合自然语言和科学语言之间的鸿沟可以让化学家用自然语言交流他们希望设计的分子功能,并通过对话找到分子结构和合成方法。
本文重点探讨了大规模语言模型在加速分子发现(分子设计和探索)方面的实用性。论文从传统的科学发现方法入手,介绍了分子生成和分子性质预测模型的组合,以及科学语言建模的工具和库。最后,论文探讨了通过聊天机器人将自然语言模型与分子发现过程相结合如何改变未来的分子设计。
加快分子发现周期有望为解决与人类生活相关的重要问题做出重大贡献,尤其是在药物发现领域。
未来的分子发现方法
分子发现是对传统科学方法的一大挑战,因为需要优化各种特性。在分子设计、制造、测试和分析(DMTA)循环中,合成分子的成本和时间限制是阻碍研究进展的瓶颈。传统方法依赖于药物化学家的 "分子假设",这种假设并不详尽,往往会延误对全球性挑战的回应。多年来,人们一直强烈要求简化分子发现过程。目前的主要挑战是如何提高基于实验室实验的 "分子假说 "的评估速度和质量。
深度生成模型已成为加速假设生成和分子发现设计的一种有前途的手段。然而,即使是这些先进的分子生成模型,也需要高效的大规模虚拟筛选方法来有效地测试假设。加速分子发现周期在 DMTA 周期中增加了一个验证循环,使许多假设能够以较低的成本得到快速评估。这一新的循环增强了设计阶段的生成模型,并确保只有有前景的假说才能真正进入合成和物理实验阶段。
分子表征
分子表征定义了模型可以利用的信息。这里的 "表示 "指的是表示分子结构和性质的方式。随着化学语言建模(CLM)技术的进步,基于文本的分子表征方式越来越受到关注。

SMILES 是一种描述分子的文本表示法,原子、键、分支、芳香度等均由特定字符串表示。这种表示法适合用于化学语言模型,因为分子可以很容易地标记化(拆分)。不过,SMILES 并非唯一,同一分子可由多个不同的 SMILES 字符串表示。这可用于数据扩展、分子特性预测和分子生成。另一方面,SMILES 也可能产生无效的表示法,为避免这一问题,我们采用了不同的处理和归一化方法。
SELFIES 是作为 SMILES 的替代方案而开发的,旨在避免生成无效的分子表征。它以保证键价有效性的规则为基础,并保留了分支长度和环的尺寸,以避免生成开放的分支和环。这可确保在生成分子时始终获得有效的表示,但可能会因为太短而无法表示有用的分子。
InChI(国际化学标识符)由国际理论化学和应用化学联合会(IUPAC)推出,是一个分层编码分子结构信息的字符串。对于大分子来说,这个字符串可能又长又复杂。为了解决这个问题,我们开发了一种名为 InChIKey 的散列,以方便搜索和检索。然而,InChI 在化学语言模型中的应用并不普遍。
这些基于文本的分子表征在训练化学语言模型方面发挥着重要作用。模型可以从这些表征中学习分子的性质和结构,并利用它们生成新分子或预测现有分子的性质。每种表征方法都有自己的优势和局限性,应根据目的选择合适的方法。
形成过程
生成模型是学习数据的基本分布以生成新样本的过程,在提高药物发现效率方面发挥着重要作用。这种技术主要有两种模型:一种是条件生成模型,利用特定的数据属性或标签生成具有理想属性的新样本;另一种是无条件模型,生成与训练数据相似的分子。条件生成模型尤其有助于以目标为导向的假设设计,大大提高了药物开发的效率。
下图举例说明了使用语言模型生成条件分子的过程。
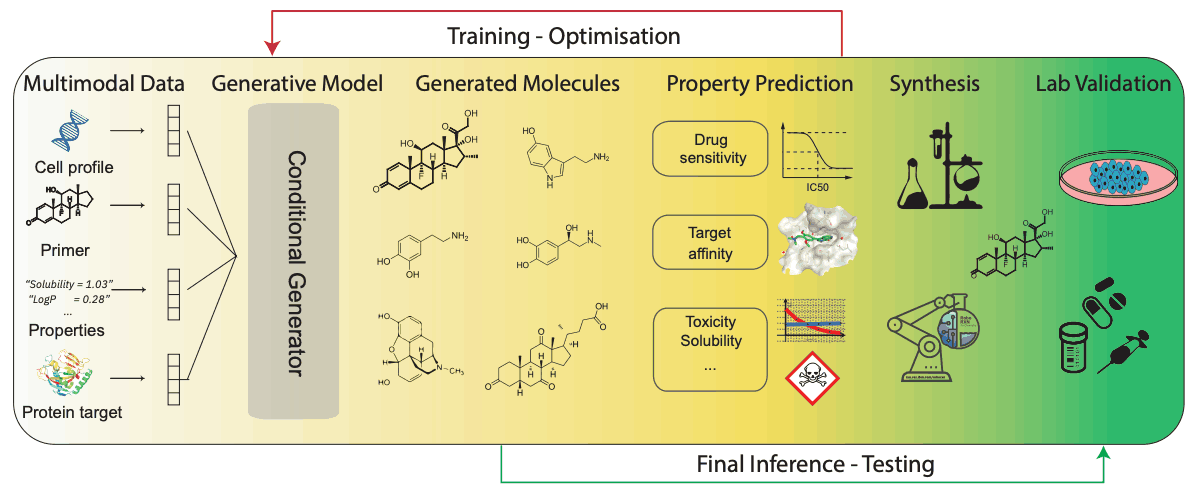
这一过程从收集和处理多模态数据开始,然后将这些数据压缩为固定大小的潜在表征。然后将这些表征传递给分子生成模型。在训练过程中,生成的分子通过反馈回路接收与生成模型相关联的内部(计算)属性预测。内部模型使用奖励函数将属性或任务驱动的分子导向生成模型。在推理阶段,优化模型生成的候选分子将继续通过实验室合成和随后的实验验证工作流程,以确定其对所需任务的效果。
介绍用于科学语言建模的先进软件工具
开源软件在科学界的发展带来了革命性的变化,尤其是在化学领域。这在许多方面促进了新研究方法的开发,提高了科学成果的可重复性。这里的重点是对分子发现有用的软件工具,从 Python 软件包到基于云的网络应用程序,不一而足。
首先是自然语言处理模型:Transformer 技术的成功被誉为自然语言处理领域的一大突破,而 HuggingFace 开发的 transformers 库的流行则进一步凸显了这一点。这些技术已被应用于计算机视觉、强化学习和蛋白质结构预测等多个领域。HuggingFace 提供的语言模型包括新模型的实现和可用于微调和推理的预训练模型。其中一些模型专门针对生命科学领域,如分子特性预测和基于文本的分子生成。
科学发现生成工具包(GT4SD)是一个旨在简化科学发现的工具包。GT4SD 支持使用、学习、微调和共享最先进的生成模型。该工具包提供了一系列技术,包括扩散和图形生成模型,使研究人员能够高效地推进有机材料设计等项目。 GT4SD 提供了一个统一的界面,易于运行和学习生成模型。它提供了一个命令行工具,并有一个模型中心,用于在云中共享学习到的模型。它还在小分子、蛋白质和晶体等领域提供了许多性质预测终点和预训练算法,并提供免费的网络应用程序和教学笔记本。
预测化学反应和确定合成路线是化学研究中的一项关键挑战。该领域最先进的技术是 IBM RXN 化学平台提供的 "rxn4chemistry "库。该工具将自然语言处理技术应用于化学领域,将化学反应视为序列转换问题,其中原子、分子和反应分别表示为字母、单词和句子。
分子转换器(MT)是 rxn4chemistry 的核心架构,采用自回归编码器-解码器模型。该模型与其他许多模型的不同之处在于,它以数据为驱动,无需使用模板即可预测化学反应的结果,并能直接表示立体化学。这使它在区域和立体选择性反应中具有很高的性能,MT 的应用范围很广,从单步逆合成到酶促反应。
在有机化学自动化方面,RXN for Chemistry 还能探索可在机器人平台(如 IBM RoboRXN)上自动执行的合成方案。它代表了化学反应建模和合成协议自动化领域的创新进步。使用编码器-解码器转换器,可以从专利实验程序中提取化学合成动作,并直接从反应 SMILES 中进行预测。这些模型还可由机器人平台通过网络接口直接控制和监测。RXN 化学平台可通过 rxn4chemistry Python 软件包访问,该软件包提供了一套丰富的语言模型rxn4chemistry Python 软件包。研究人员可以自由访问该软件包,并将其用于不同的化学反应任务。它可用于多步逆向合成分析计划等复杂任务,并包括不基于 Transformer 的模型。
除此之外,HuggingMolecules 还有一个专门用于聚合、标准化和分发分子特性预测语言模型的库。其中有一些仅编码器的 CLM,带有几何和结构识别偏差(如 MAT 及其后继 R-MAT),以及基于 SMILES 训练的纯 BERT 模型(如 MolBERT 和 ChemBERTA)。
rxn-chemutils 是一个库,包含 RXN for Chemistry 的化学相关实用程序,包括 SMILES 标准化功能(如规范化和消毒)以及与其他表示法(如 InChI)的转换功能。)的转换功能,包括以下内容。它协调反应 SMILES 并为 CLM 的使用做好准备,还包括 SMILES 的扩展(如以非规范化顺序遍历分子图)和标记化。另一个具有类似功能的库是 pytoda。它也支持不同的语言(如 SELFIES 和 BigSMILES)和标记化方案(如 SMILES-PE)。蛋白质也有类似的功能,包括不同的语言(IUPAC、UniRep、Blosum62)和蛋白质序列扩展策略。对于小分子、蛋白质和聚合物,专用的语言类有助于与 LM 集成、存储词汇表、执行在线转换和输入自定义数据集。数据集可用于预测小分子、蛋白质和聚合物的分子特性、药物敏感性、蛋白质配体亲和性或自我监督。
分子发现的未来
直到几年前,利用人工智能模型进行科学知识提取和计算分析的想法还是一个雄心勃勃的梦想,就像想象搜索引擎的存在一样。科学思维的核心是推理能力,而人工智能以与人类相同的方式进行推理的那一天尚未到来。不过,人工智能可以学习和模仿人类行为:ChatGPT 和 GitHub Copilot 等大型语言模型就是在我们记录的大量数据基础上训练出来的。如果将其应用到计算科学领域,非专业人员也能通过精心设计的提示自信地进行计算分析。这一过程允许科学家为模型提供反馈并对其进行优化。这将使非科学背景的人更容易进行科学探索,因为他们无需专业培训就能进行科学分析。这一发展为分子发现领域的新革命打开了大门。未来,类似聊天机器人的界面将负责所有计算过程。这种界面将支持分子发现的整个过程,从设计构思开始,到合成规划、材料采购、定期安全检查和实验验证。
传统上,为特定任务专门训练的神经网络需要开发新的模型来应对新的任务。然而,最近在大规模语言建模方面取得的进展正在从根本上改变这种方法。现在,"基础模型 "通过在庞大的数据集上进行训练,已经能够执行多种任务。这为自然语言处理领域开辟了新的研究方向,如提示工程和语境学习。
化学领域也正在引入基础模型。结合自然语言和化学语言的特定任务模型正在开发中,而结合性质预测、反应预测和分子生成的多任务模型也正在出现。这些模型通过支持从自然文本到发现新分子、提出合成途径和执行实际合成协议的整个过程,显示出优于传统模型的性能。
这些进步极大地促进了科学探索和技术创新的加速。分子发现领域的未来进展也备受期待。
整合化学工具和聊天机器人
鉴于大规模语言模型的强大多功能性,围绕它们构建聊天机器人界面是一个自然的发展过程;许多类似的工具已经出现,如 ChatGPT。这些工具在简单的化学任务中表现出色,让化学家可以与化学数据交互,解决化学任务。此外,计算机科学家为药物发现和材料科学开发的模型也可通过大规模语言模型使用。这使得不具备使用这些人工智能模型所需的编程技能的专家也能轻松获得最新技术。
通过将聊天机器人与 PubChem、RDKit 和 GT4SD 等现有化学软件工具集成,可以为聊天机器人提供便利。这些应用可以提高这些模型的使用率,最大限度地发挥其潜力和价值。下图展示了使用本文构建的 ChemChat 聊天界面使用各种化学工具的示例。
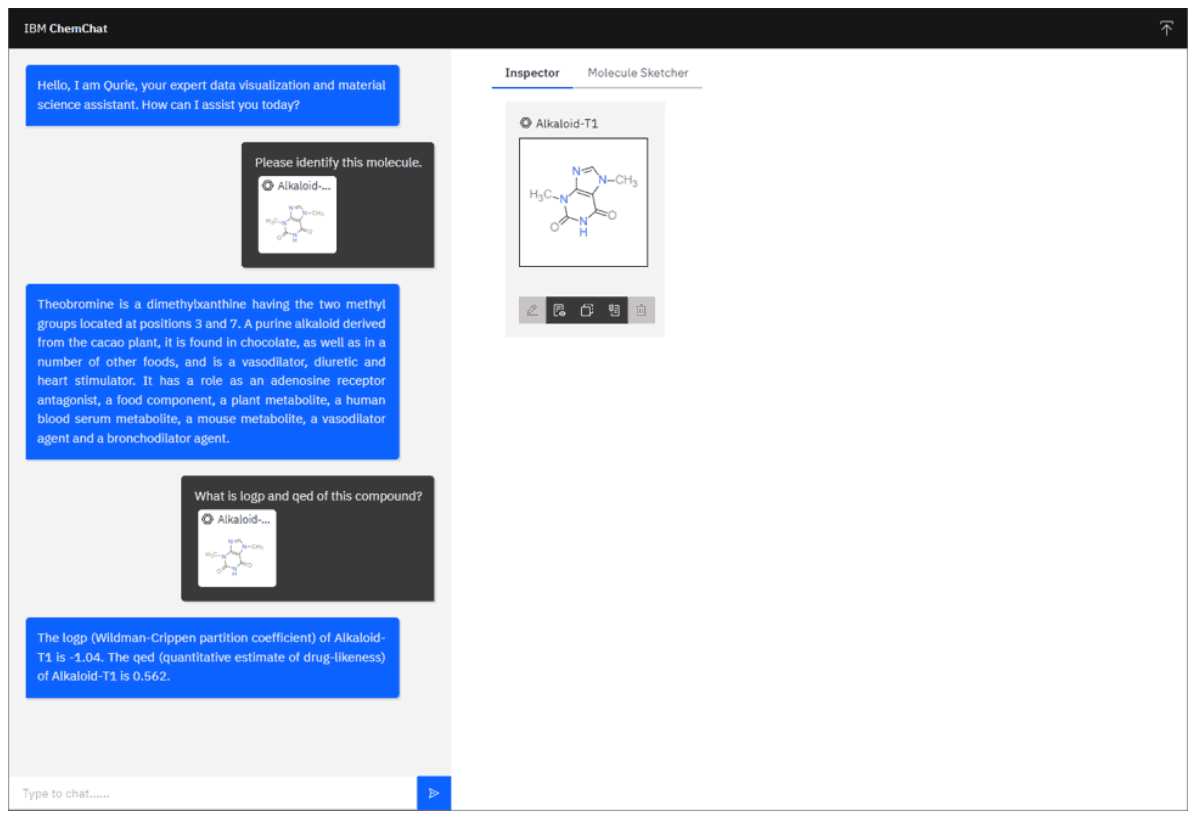
在这个例子中,用户首先提供了一个分子结构,并被要求识别该分子。用户输入的信息会被发送到大规模语言模型,如果认为 PubChem 等支持工具能够回答问题,聊天机器人就会向 PubChem API 发送请求,PubChem API 会返回分子的简要描述。然后会要求用户计算 logP 分布系数和类药物特性定量估计值 (QED)。这些性质的计算通过 GT4SD 工具完成,并由聊天机器人回答。
将现有工具与大规模语言模型相结合,可创建一个用于材料科学和数据可视化的聊天机器人助手,它可以执行简单的编程任务,而用户无需了解编程或访问计算资源。

在 ChemChat 中,当有三个候选 SMILES 在回答这个问题时被提及时,经过一些后处理,文本结果被可视化。经过后处理后的结果可视化。
聊天机器人非常有用,这一点从 ChatGPT 的迅速发展中可见一斑。通过利用大规模语言模型,可以轻松进行复杂的化学信息处理。现有化学工具和自然语言能力之间的协同作用有可能彻底改变化学研究和实验的方式。
总结
在化学领域,即使随着功能分子和生物化学研究的发展,复杂的分子设计仍然需要大量的时间和高昂的费用。然而,大规模语言模型的使用有可能大大加快分子发现的速度。这些语言模型有可能学习化学结构,并通过解释和生成文本高效地探索化学搜索空间。
将预测分子生成和特性的模型、科学语言建模和使用最先进的软件工具结合起来,可以推动分子发现的加速发展。 基于文本的分子表征,如 SMILES、SELFIES 和 InChI,可以提供新分子的生成和特性。有条件的生成建模可以创造新的分子。
此外,以 Python 软件包和网络应用程序为形式的开放源码软件的发展也为促进分子发现提供了工具。预测化学反应、确定合成路线以及将大规模语言模型与化学工具相结合,是塑造未来分子发现的关键因素。
这些进步有望加速与人们生活息息相关领域的科学探索和创新,包括药物发现。聊天机器人界面以大规模语言模型为核心,与化学信息学软件无缝集成,方便没有专业知识的任何人进行科学分析,它的出现预示着分子设计和发现领域的一场新革命。