一、写在前面
最近发现一篇"有点意思"的文章,来自北京大学口腔医学院、国家口腔医学中心的作者挖掘整合分析了多个GEO数据的微阵列、Bulk RNA-seq通过四种机器学习算法筛选了159个核心DEGs后通过GEO中的scRNA-seq、空转数据挖掘进一步确定SASH1的细胞与空间定位情况,补了六个样本的细胞系WB实验后,就水灵灵的在25年12月发在了《INTERNATIONAL JOURNAL OF SURGERY》这一IF=10.1的期刊上。
Dai Z, Shan X, Kang Y, et al. A multi-omics pipeline integrating machine learning and spatial-cellular analysis identifies SASH1 as a prognostic biomarker and therapeutic target in head and neck squamous cell carcinoma. Int J Surg. 2025 Dec 1;111(12):9178-9195.
doi: 10.1097/JS9.0000000000003647.
原文链接:https://pmc.ncbi.nlm.nih.gov/articles/PMC12695206/
本文用到的很多生信分析/实验技能我们也分享过教程,可参考:
二、背景介绍
头颈鳞状细胞癌(HNSCC)是一种常见恶性肿瘤,起源于口腔上皮组织、鼻咽、咽下、喉部及其他上气道消化道区域,占所有头颈部肿瘤的90%以上。高通量测序的广泛应用极大地推动了肿瘤分子机制的研究。公共数据库,如基因表达全集(GEO)和癌症基因组图谱(TCGA),汇总大量转录组数据,支持跨数据集分析,提升研究结果的稳健性。作者指出,仍存在若干局限性。首先,许多研究依赖单一数据集,这可能导致样本和平台特异性偏差导致不可重现的结论。其次,虽然一些研究利用机器学习进行特征选择,但单一算法的使用可能存在模型偏差或过拟合,可能导致无法识别具有生物学意义的分子。更重要的是,即使是从大批量数据中识别出的稳健生物标志物,也缺乏细胞和空间分辨率。目前尚不清楚基因的差异表达是源自癌细胞本身,还是源自TME的变化,以及其表达如何在组织结构中组织。这一知识空白限制了我们对机制的理解以及许多假定生物标志物的临床转化性。
为解决这些多方面的局限性,该研究建立(挖掘)了全面的多组学流程,系统识别和验证具有高诊断和预后潜力的HNSCC分子生物标志物。作者整合了多个公共数据库(GEO和TCGA)的转录组数据,并采用了一套互补的机器学习算法,对核心候选基因进行了稳健筛选。关键是,为了克服体分析的局限,作者利用单细胞和空间转录组学,剖析了这些核心基因在HNSCC生态系统中的细胞起源及原位拓扑模式。最后,通过Western blot实验验证了最终关键候选物SAM和SH3结构域1(SASH1)的蛋白质级表达。通过构建跨越大规模计算筛选到高分辨率细胞、空间和蛋白质层面验证的多层证据链,本研究不仅旨在识别新颖可靠的生物标志物,还旨在提供更深层次的机制性见解。这些发现对推动精准医疗的发展和改善HNSCC患者的临床结果具有希望。
三、主要结果
为了不断章取义,我们几乎以直译的方式进行分分享:
1、差异分析
作者通过DEA在GSE29330数据集中共识别了1599个DEGs,其中包括1224个下调基因和375个上调基因(见图 1a,b)。对GSE6631数据集的分析显示,873个DEGs,包括384个下调基因和489个上调基因(图 1 c,d)。由于不同数据源可能产生批量效应,GSE29330和GSE6631进行了批量效应校正和数据整合,以确保后续分析的可靠性和一致性。图1e,f展示了批次校正前后样本表达分布的箱型图,在校正前观察到两数据集表达分布的显著差异,校正后实现比对。PCA进一步验证了校正,显示校正前样本分离清晰,校正后分布均匀混合(图 1g,h)。使用批量校正数据集,重复DEA,识别出190个DEGs(118个下调,72个上调)(图 1i,j),为后续功能富集分析和生物学解释奠定了基础。
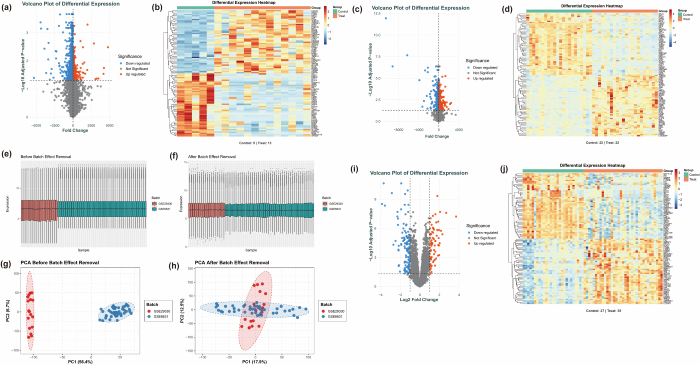
图1
2、GO和KEGG对重叠基因的分析
为探讨与HNSCC相关的DEGs的生物学功能,作者于TCGA-HNSC数据集中识别了1319个DEGs(549个上调,770个下调),如火山图所示(图 2a)和基于调整后P值的基因排序点图(见图 2b)。这些与批次校正数据集中的190个DEG相交,产生159个重叠DEG,如图2c所示。对这159种DEGs的Go分析显示,与ECM重塑相关的BP显著富集,包括ECM组织、胶原代谢过程和外部包覆结构组织。对于CC,DEG主要与含胶原的ECM、肌节和胶原三聚体相关。多重分子富含ECM结构成分、丝氨酸型内肽酶活性和整合素结合,表明其在ECM稳定性和细胞间信号传导中起关键作用(见图 2d)。KEGG分析发现了与肿瘤发生相关的通路显著富集,包括ECM受体相互作用、局部粘附、PI3K-Akt信号传导以及肌肉细胞中的细胞骨架调控。此外,AGE-RAGE信号通路和IL-17信号通路显示显著富集,表明其可能与炎症和代谢失调有关(见图 2 e,f)。KEGG通路相关热图进一步揭示了ECM相关通路、PI3K-Akt和松弛素信号通路之间的功能重叠与协同(见图 2e)。这159个重叠的DEGs合计可能通过调控ECM动态、细胞粘附和关键信号通路,在HNSCC进展中发挥关键作用。
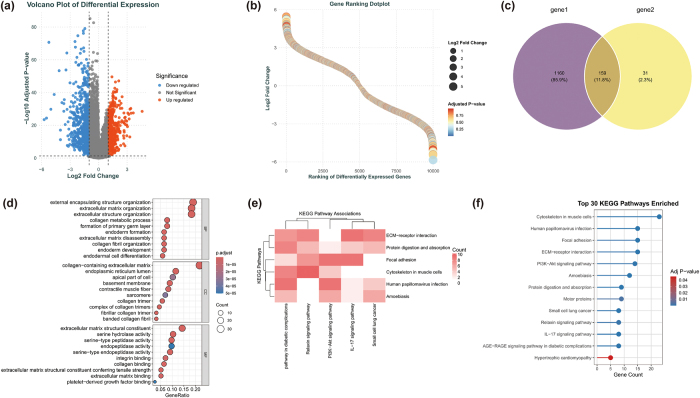
图2
3、多机器学习算法集成及关键核心基因的独立验证
为了识别与HNSCC相关的159个DEG中的关键预测特征,作者系统地应用了四种特征选择方法------LASSO回归、SVM-RFE、XGBoost和Boruta,分别利用线性建模、支持向量学习、集合树模型和随机森林稳定性选择。LASSO回归对标准化的159个DEG进行了十倍交叉验证,确定了最优正则化参数λ(图 3a,b)。带有最小均方误差(MSE)的交叉验证曲线,用橙色虚线标示,表示偏置与方差之间的最佳平衡(图 3a)。系数路径显示,随着λ增加,基因系数趋近于零,18个基因在最优λ下仍保持非零系数,表明预测潜力较强(图 3b)。SVM-RFE评估了模型在不同特征计数下的表现(见图 3c,d))。随着特征数增加,泛化误差递减如图3c所示,18个特征时峰值CV准确率(CV准确率=0.905)和最小误差(CV误差=0.0952)验证了该特征子集的优越性(图 3d)。XGBoost基于梯度提升分析了159个DEG(图 3e--g)。SASH1被突出为增益最高的特征,其次是EMP1、GPD1L、KRT4和MGLL(见图 3e)。SASH1在样本中的广泛影响,EMP1、GPD1L和KRT4也持续贡献,见图3 f)。决策树结构确认SASH1是驱动初始分裂的根节点(增益=34.18),其次是ADH1B和CDH3,产生20个高增益基因(图 3g)。Boruta利用随机森林和阴影特征,根据Z分数将基因分为被选中(绿色)、暂定基因(蓝色)和拒绝基因(红色)(见图3 h,i))。选择基因Z分数显著更高的密度分布产生了81个统计学显著特征(图3 i)。所有方法特征的交叉(LASSO:18,SVM-RFE:18,XGBoost:20,Boruta:81)发现了四个核心基因始终被选中:COL1A1、EMP1、MYH11和SASH1(图 3j)。为验证其稳健性和普遍性,在独立验证数据集GSE138206(不包括训练数据集GSE6631和GSE29330)上进行了单基因表达分析。箱形图显示SASH1(P < 0.001)、MYH11(P < 0.001)、EMP1(P < 0.001)和COL1A1(P < 0.001)的表达差异显著(见图 3k--n)。
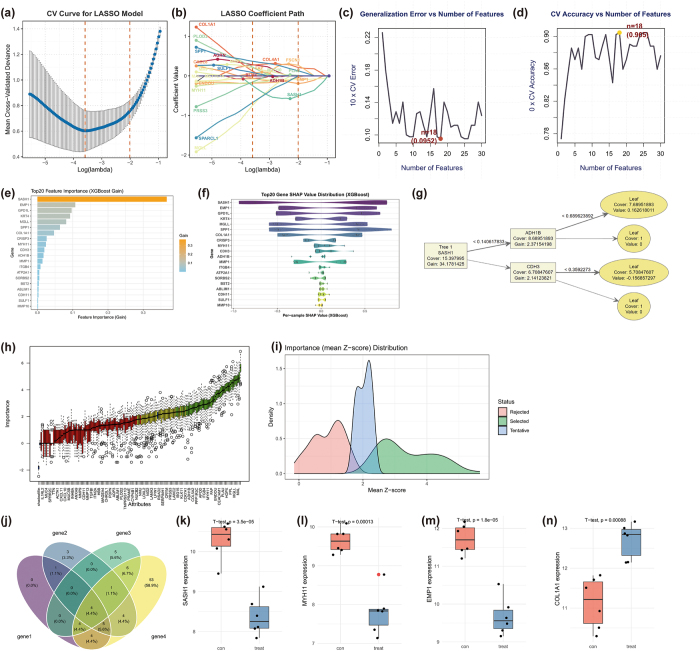
图3
4、通过单细胞分析HNSCC微环境中核心基因的细胞异质性
为解析四个核心基因(COL1A1、EMP1、MYH11、SASH1)的表达谱,分析了公开的HNSCC单细胞转录组数据。通过无监督聚类和随后的人工注释,共识别出11种主要细胞类型,包括恶性细胞、成纤维细胞、内皮细胞,以及多种免疫细胞如T细胞、B细胞、浆细胞、巨噬细胞、NK细胞、树突状细胞和肥大细胞。这些细胞共同构成肿瘤微环境的细胞图谱(图 4 a)。对四大核心基因在所有细胞类型中的表达水平和百分比的全面分析揭示了明显的模式(见图 4b)。COL1A1表达在成纤维细胞簇(c-6、c-19)中高度集中,平均表达最高,阳性率超过75%。EMP1分布更广,在成纤维细胞(c-6、c-19)、部分恶性细胞簇(c-8)和内皮细胞(c-10、c-18)中检测到中等表达水平。MYH11在所有已知细胞类型中均表达极低,阳性细胞比例极低。与其他细胞不同,SASH1的表达主要在间质细胞和免疫细胞中观察到,平均表达在成纤维细胞、内皮细胞、B细胞和浆细胞中更高。相反,在多个标记为恶性细胞的亚群中,其平均表达水平相对较低(如c-1、c-3、c-4、c-6、c-8、c-13、c-15、c-20)。为了进一步说明每个基因的表达分布,生成了特征图和小提琴图(图 4c-j)。SASH1的特征图直观地显示,其表达信号(暖色)主要分布在非恶性细胞区域,而在大型恶性细胞区域则呈现为低表达的"冷区"(见图 4c)。相应的小提琴图确认,SASH1在所有恶性细胞簇中的中位表达接近零(见图4 d)。MYH11在所有细胞类型中保持最低基线表达(见图4 e,f)。EMP1表达在多个簇中被检测到,其峰值出现在c-6(成纤维细胞)和c-8(恶性细胞)(见图4 g,h))。COL1A1的表达模式最为特异,其高表达信号几乎仅限于两个成纤维细胞簇C-6和C-19,而在其他细胞类型中几乎为零(见图 4 i,j。此外,为了研究这些核心基因在恶性细胞群体中的表达动态,构建了细胞轨迹(见图5 a)。通过根据该群体中位表达将恶性细胞分层为高表达组和低表达组后,可视化显示了四个基因的不同动态模式。对于SASH1和MYH11,几乎所有恶性细胞都被归类为低表达状态(蓝色),只有少数细胞表现出高表达(见图5 b,c)。相比之下,EMP1和COL1A1在恶性细胞群体中表现出明显的表达异质性,既有高表达(粉色)和低表达(蓝色)亚群。值得注意的是,EMP1高细胞聚集在轨迹图左上角的特定分支上(图5 d),而COL1A1高细胞也表现出聚集分布,主要分布在轨迹左侧和右上方的两个小分支(图 5e)。
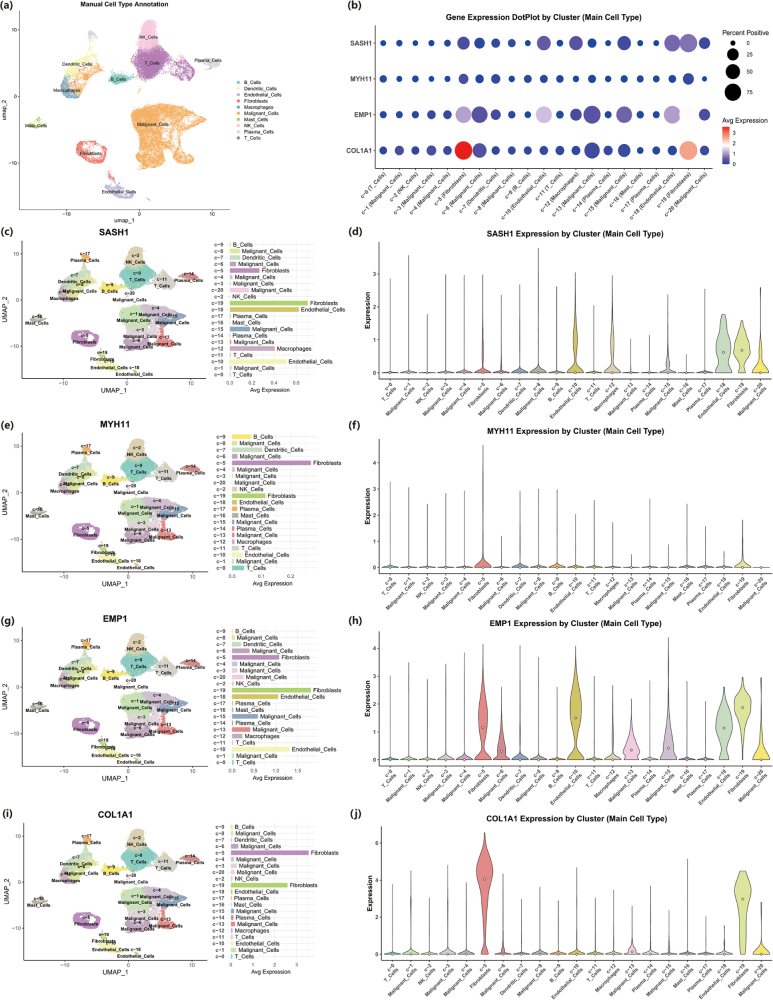
图4
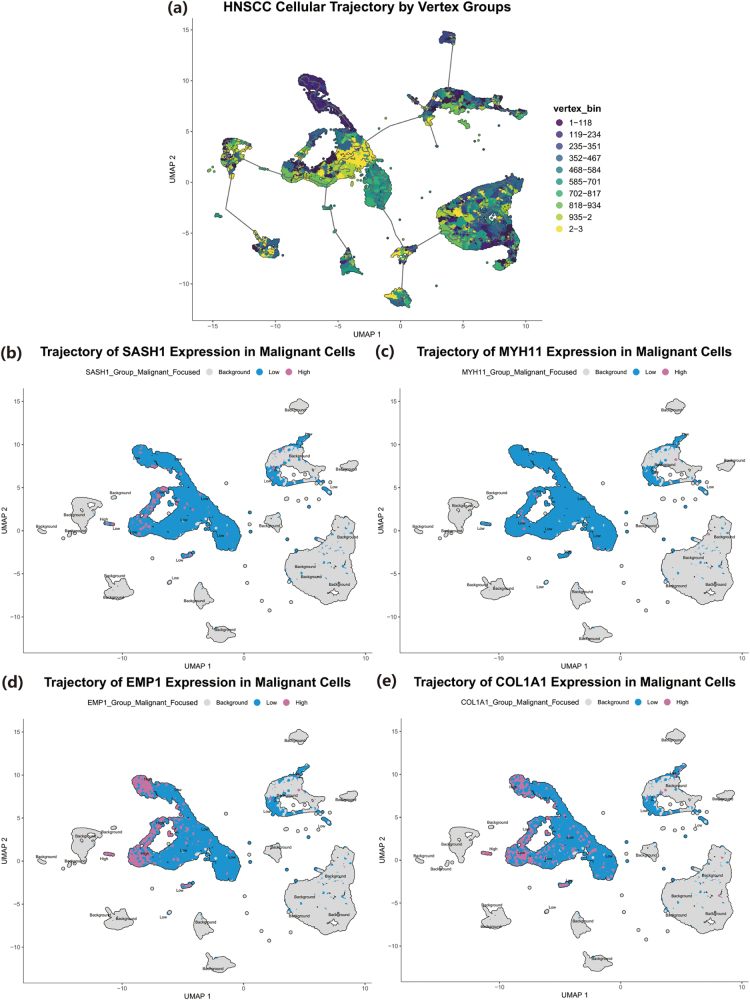
图5
5、HNSCC组织核心基因的空间拓扑
为了研究四个核心基因在原位组织环境中的空间定位,对具有代表性的HNSCC样本进行了空间转录组学。该样本的H&E染色展示了其组织病理学形态,显示了致密的肿瘤细胞巢穴及周围的基质结构(图6 a)。在对空间转录组谱进行无监督聚类后,所得的空间域根据关键标记基因的表达模式被人工标记为六个功能上不同的区间。这些划分清晰地界定了组织内的功能异质性,主要包括纤维化间质区、富含髓系的炎症区,以及四个不同的肿瘤亚区:分化/角化肿瘤、代谢/分泌肿瘤、基底样增殖肿瘤和类癌干细胞(CSC样)生态位(图6 b)。在这份注释空间图谱上,四个核心基因的表达被可视化,揭示了截然不同且高度结构化的空间分布模式。SASH1在绝大多数组织中表达显著偏低,尤其是在标注为纤维性纤维质的大面积致密区域及大多数肿瘤亚区域内。其表达信号在整个组织切片中几乎均匀呈冷色调(低表达),表明其在既定肿瘤生态系统中总体上处于下调状态(见图6 c)。
与SASH1形成鲜明对比的是,COL1A1的表达清晰地描绘了肿瘤的基质结构。其高表达信号(暖色)几乎完全且强烈地在标注为纤维化间质的区域中丰富。相反,其在所有肿瘤亚区域和髓系炎症区的表达都很低。这一结果生动地展示了纤维化微环境,并突出COL1A1高纤维化基质与SASH1低肿瘤区之间明显的空间排斥模式(见图 6d)。另外两个核心基因也展现出独特的空间生态位。EMP1的高表达信号显著定位于肿瘤-基质界面,尤其集中在直接毗邻纤维化基质的分化/角化肿瘤区和代谢/分泌性肿瘤区。这一模式表明EMP1可能在上皮肿瘤成分与周围纤维基质之间的相互作用中发挥作用(见图 6e)。MYH11的表达通常较低,且在组织中更为弥漫。虽然其信号在纤维化间质部分的检测度略高于肿瘤核心,但未表现出其他基因所观察到的强烈特异性富集模式(见图6f)。总之,这些结果详细描绘了HNSCC肿瘤微环境中四个核心基因的细粒度空间组织。
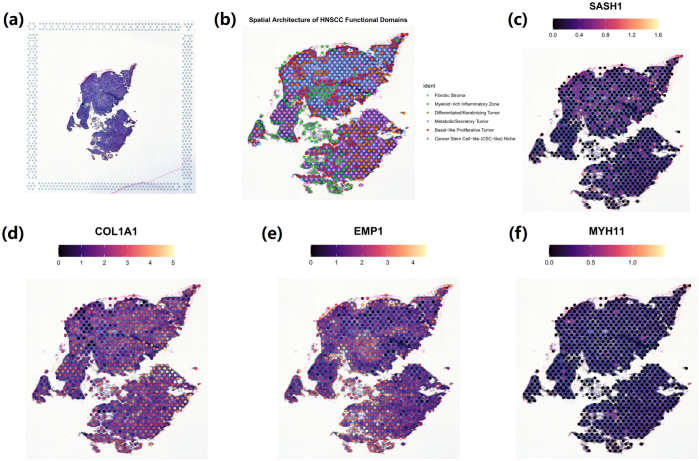
图6
6、HNSCC关键基因的表达差异及预后价值
为进一步研究HNSCC中四个关键基因(COL1A1、EMP1、MYH11和SASH1)的表达差异及生物学意义,采用TCGA-HNSC队列进行了单基因表达分析(见图7 a-d)。肿瘤与正常组织之间四个基因均有显著表达差异(P < 0.05)。肿瘤组织中COL1A1和EMP1显著上调,而MYH11和SASH1则显著下调。患者根据中位基因表达分为高表达组和低表达组,并生成了Kaplan-Meier生存曲线(见图7 e-h)。SASH1表达与预后呈相关趋势(P = 0.0463),低表达组的生存概率较低,表明SASH1可能成为有利的预后标志,但尚需进一步验证。相比之下,COL1A1(P = 0.710)、MYH11(P = 0.331)和EMP1(P = 0.984)在高表达组和低表达组间的生存率上无统计学显著差异,表明其表达可能不会直接影响整体生存期,或需要更大样本量进行进一步验证。
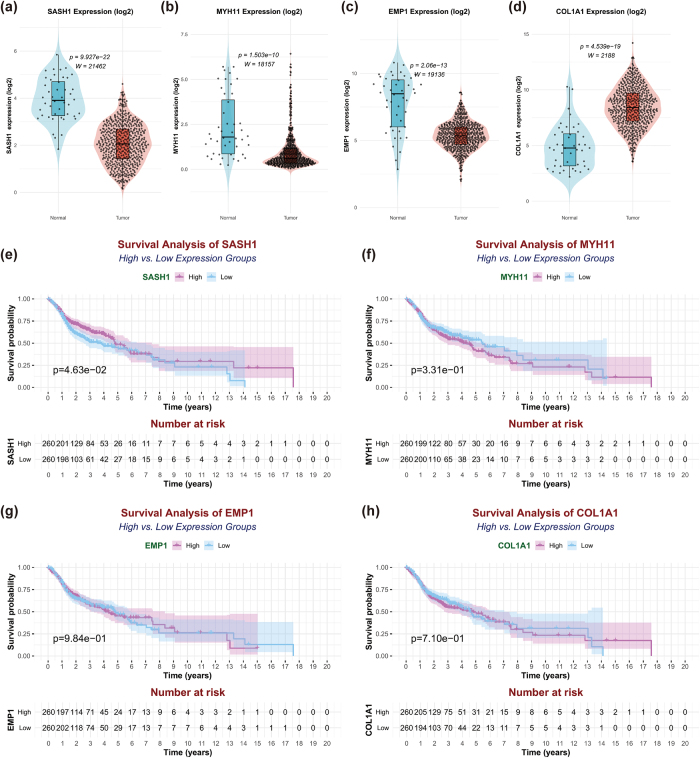
图7
7、SASH1相关基因的功能注释与途径富集分析
为了探究SASH1的生物学功能,Spearman相关分析发现了与SASH1表达显著相关的基因(见图 8a-c)。与FAM214A(r = 0.64,P = 2.6e−60)、SLC16A7(r = 0.60,P = 6e−53)和STXBP5(r = 0.62,P = 3.76e−56)均有强烈 正相关,表明功能协同效应。 对相关基因集进行了GO和KEGG富集分析,以探讨双相位障碍和双重性脂肪及富集通路(见图 8d,e))。研究发现了有丝分裂核分裂、小脑浦肯野细胞分化和细胞粘附调控等过程的显著富集,以及分子功能如小GTP酶结合和核糖核蛋白复合物结合(见图8d)。检测出包括甲状腺激素信号传导、切迹信号传导、核苷酸代谢、孕酮介导的卵母细胞成熟、卵母细胞减数分裂、泛素介导蛋白水解和紧密连接等通路的富集(图8e)。这些通路在细胞分裂、分化、代谢调控和信号传导中至关重要,支持了SASH1在调控多种细胞功能中发挥重要作用的假说。
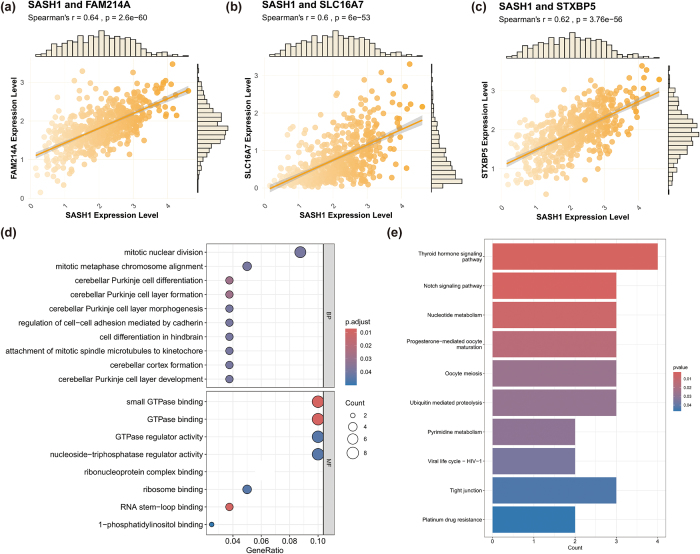
图8
8、SASH表达与临床特征及药物敏感性的关联分析
为评估SASH1的临床相关性,分析了其表达与临床特征的关联性(见图。9(a-e))。热图和箱形图显示不同年龄组间无显著表达差异(P = 0.76),表明与年龄无关联。然而,SASH1表达差异在生存状态(P = 0.025)、性别(P = 0.038)和临床阶段(P = 0.01)方面具有统计学显著性,表明与预后、性别差异及肿瘤进展有关。药物敏感性分析显示SASH1表达与药物反应之间存在显著相关性(见图。10(a-i))。一个火山图显示ZM447439与SASH1表达式(r ≈ −0.5,P < 1e−30)呈负相关性最强( 见图。10(a))。箱形图和散点图证实,高SASH1表达与ZM447439敏感性降低相关(R = −0.52,P < 2.2e−16)(图。 10(b-c))。同样,KU-55 933也与SASH1表达式呈负相关(R = −0.48,P < 2.2e−16)(图 。10(e, h)),进一步支持SASH1在药物耐药中的潜在作用。相反,AZD7762和AZD6738与SASH1表达呈正相关,表明高SASH1表达样本的药物敏感性增强(R = 0.47和R = 0.44,P < 2.2e−16)(见图。 10(d、g、f、i))。这些发现凸显了SASH1在精准医疗中的潜在价值,因为它可能影响对多种药物的敏感性,从而指导个性化治疗策略。
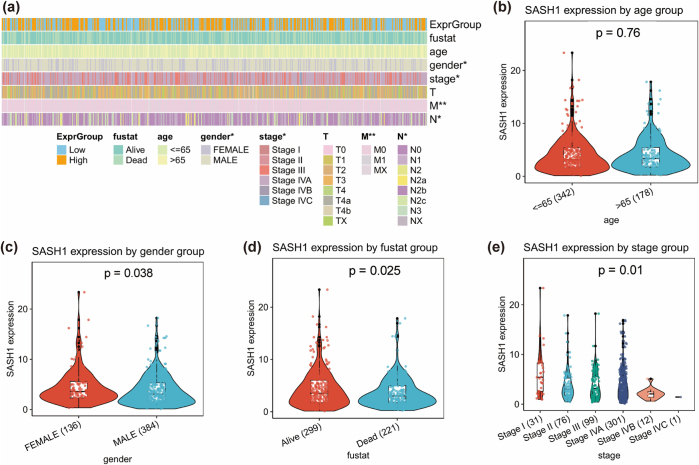
图9
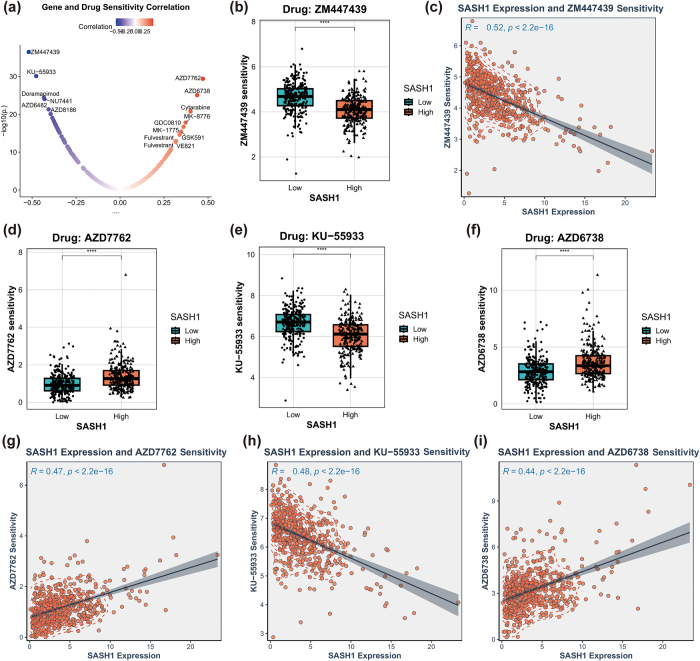
图10
9、Western印迹验证确认SASH1在蛋白质水平下调
为验证HNSCC蛋白层面SASH1的表达变化,进行了Western印迹分析。结果显示,OSCC组SASH1蛋白表达水平与对照组显著变化。具有代表性的Western印迹带清楚地表明,在对照组细胞中检测到一条约137 kDa的SASH1蛋白带,而OSCC组细胞中该带强度显著降低。内部对照组β-肌动蛋白(~42 kDa)的表达在两组之间保持稳定,表明蛋白质负荷相等(见图)。11(a))。为量化该结果,对三项独立实验的带状物进行了密度测量分析。在归一化为β-肌动蛋白后,相对蛋白表达水平显示,OSCC组的SASH1蛋白水平显著低于对照组(见图 11b,P < 0.01)。
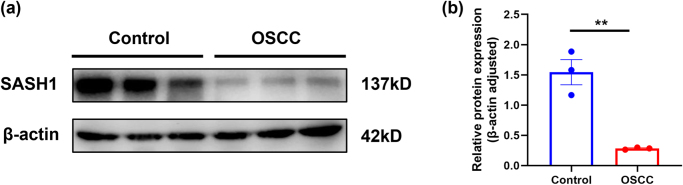
图11
最后聊聊
可以看出作者还是比较全面,RNA-Seq、微阵列、单细胞、空转、机器学习、WB实验样样都来。很多内容我们都分享过,相信这些技能大家也都会,至于能不能发10分以上的文章吗,这个事情就很复杂了。
本文用到的很多分析/实验技能我们也分享过教程,可参考: