文章目录
- 背景
- 部署过程
-
- 1,确保curl、git、wget工具已安装
- [2,确保CUDA compiler driver >=11.8](#2,确保CUDA compiler driver >=11.8)
- [3,按照对应指引安装 pixi 包管理器](#3,按照对应指引安装 pixi 包管理器)
- 4,安装localcolabfold
- 5,脚本示例演示
- 6,脚本示例输出结果解读
- 7,其他的演示例子
背景
想要自主、自由地进行蛋白质结构预测是一件比较奢侈的事,毕竟像AlphaFold之类的主流蛋白质折叠(也就是结构预测)model不是谁都有那个硬件设备以及算力去部署的;
如果是在超算之类的平台上部署,那么用又成了另外一个问题:首先是要收费,课题组开销不一定能够划分出来这一块;其次是超算系统操作毕竟没有本地舒服自在,还是比较麻烦的,得适应一下。
而且正是因为在超算之类的公家平台上做什么都得收费,所以debug得千万小心,而且一定得做小测试,要不然一个debug小问题,就会烧掉一大笔钱。

我们一般预测结构,或者说想获取结构,也就那么几个来源:
- 预测天然的、简单的蛋白质:
- PDB、AFDB数据库有的,就不用自己去预测,直接有现成的
- 数据库没有的,我们再试试一些结构预测软件,比如说AlphaFold3
- 一些结构预测软件没有那个实力部署的,比如说AlphaFold3只能平时白嫖网页端,那可以看一下ColabFold,这个我之前提过很多次了,只要自己有一些简单的算力基础
- 预测更复杂的任务:复合物、突变,并且想要批量处理等等
- 能部署AlphaFold3之类的去部署
- 没有那种硬件设置以及算力的,向下用ColabFold
但是google colab平台上的colabFold毕竟用的是google家的算力,一旦扯到gpu又得充值收费,而且设置起来也不方便。
我们想用结构预测的需求就是完全自由、尽可能的自由,就是随时随地进行部署。
所以我这里推荐本地部署ColabFold(https://github.com/YoshitakaMo/localcolabfold)
部署过程
1,确保curl、git、wget工具已安装
如果没有安装,可以通过以下命令安装
markdown
sudo apt -y install curl git wget2,确保CUDA compiler driver >=11.8
确保你的Cuda编译器驱动程序是11.8或更高版本(最新版本12.4更佳)。如果你没有GPU或者不打算使用GPU,你可以跳过这一步。
可以通过以下命令查看
markdown
nvcc --version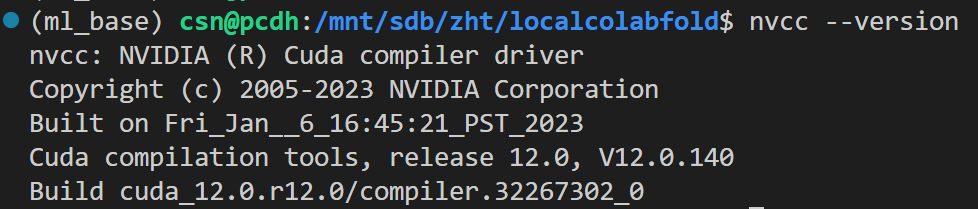
3,按照对应指引安装 pixi 包管理器
markdown
curl -fsSL https://pixi.sh/install.sh | sh参考:https://pixi.prefix.dev/latest/installation
4,安装localcolabfold
markdown
git clone https://github.com/yoshitakamo/localcolabfold.git
cd localcolabfold
pixi install && pixi run setuplocalcolabfold就安装在localcolabfold/.pixi/envs/default文件夹中
5,脚本示例演示
安装好之后有一个演示脚本run_colabfoldbatch_sample.sh,
markdown
#!/bin/bash
INPUTFILE="1BJP_1"
OUTPUTDIR="${INPUTFILE}"
RANDOMSEED=0
export PATH="/path/to/localcolabfold/.pixi/envs/default/bin:${PATH}"
colabfold_batch \
--num-recycle 3 \
--amber \
--templates \
--use-gpu-relax \
--num-models 5 \
--model-order 1,2,3,4,5 \
--random-seed ${RANDOMSEED} \
${INPUTFILE}.fasta \
${OUTPUTDIR}首先需要修改一下上述shell脚本中设置环境变量PATH的地方:
就是自己安装localcolabfold的地方,此处我按照自己的需求修改:
markdown
export PATH="/mnt/sdb/zht/localcolabfold/.pixi/envs/default/bin:${PATH}"修改注释如下,习惯用zsh的用zsh即可
markdown
#!/bin/bash
INPUTFILE="1BJP_1" # 输入文件, 不包含扩展名, 此示例中为1BJP_1.fasta
OUTPUTDIR="${INPUTFILE}" # 输出目录, 可以与输入文件相同, 也可以不同
RANDOMSEED=0
export PATH="/mnt/sdb/zht/localcolabfold/.pixi/envs/default/bin:${PATH}"
colabfold_batch \
--num-recycle 3 \
--amber \
--templates \
--use-gpu-relax \
--num-models 5 \
--model-order 1,2,3,4,5 \
--random-seed ${RANDOMSEED} \
${INPUTFILE}.fasta \
${OUTPUTDIR}- --num-recycle 3: 指定预测过程中循环迭代的次数为3次,循环迭代是一种优化策略,通过多次迭代预测,可以逐步改进预测结果的质量。增加迭代次数可能会提高预测的准确性,但同时也会增加计算时间和资源消耗
- --amber: 使用AMBER软件包进行结构优化(也称为能量最小化或结构松弛),AMBER是一种广泛使用的分子模拟软件,能够对预测的蛋白质结构进行优化,使其更加稳定和合理。通过能量最小化,可以减少结构中的不合理构象,提高预测结果的质量
- --templates: 表示在预测过程中使用模板结构,模板结构是指已知的蛋白质结构,这些结构可以作为参考,帮助预测未知蛋白质的结构。通过使用模板,可以提高预测的准确性和速度,特别是对于与模板结构相似的蛋白质
- --use-gpu-relax: 指定在进行结构优化(AMBER能量最小化)时使用Nvidia GPU加速,如果机器上安装了Nvidia GPU并且安装了相应的CUDA驱动程序,使用GPU可以显著加快结构优化的速度,提高计算效率
- --num-models 5:指定生成5个预测模型,ColabFold会根据输入的蛋白质序列生成多个预测模型,每个模型可能略有不同。通过生成多个模型,可以评估预测结果的可靠性和多样性,选择最佳的预测结果。类似于alphafold3一样,默认1个seed生成5个预测model
- --model-order 1,2,3,4,5: 指定模型的生成顺序,按照1、2、3、4、5的顺序生成5个模型
- --random-seed ${RANDOMSEED}: 设置随机种子,随机数种子在预测过程中用于初始化随机数生成器,不同的种子值可能会导致不同的预测结果。通过设置固定的种子值,可以确保预测结果的可重复性,即在相同的输入和参数下,每次运行都会得到相同的结果。我一般习惯都设置为0,因为之前在网页版默认seed用惯了,有时会设置为年份
运行的话就很简单,直接1个bash命令即可,可以tmux或者nohup挂着
markdown
bash run_colabfoldbatch_sample.sh其实我们可以看到,命令行非常简单,其实本质就只有colabfold_batch这个命令,参数只需要input的fasta序列、输出文件夹即可。
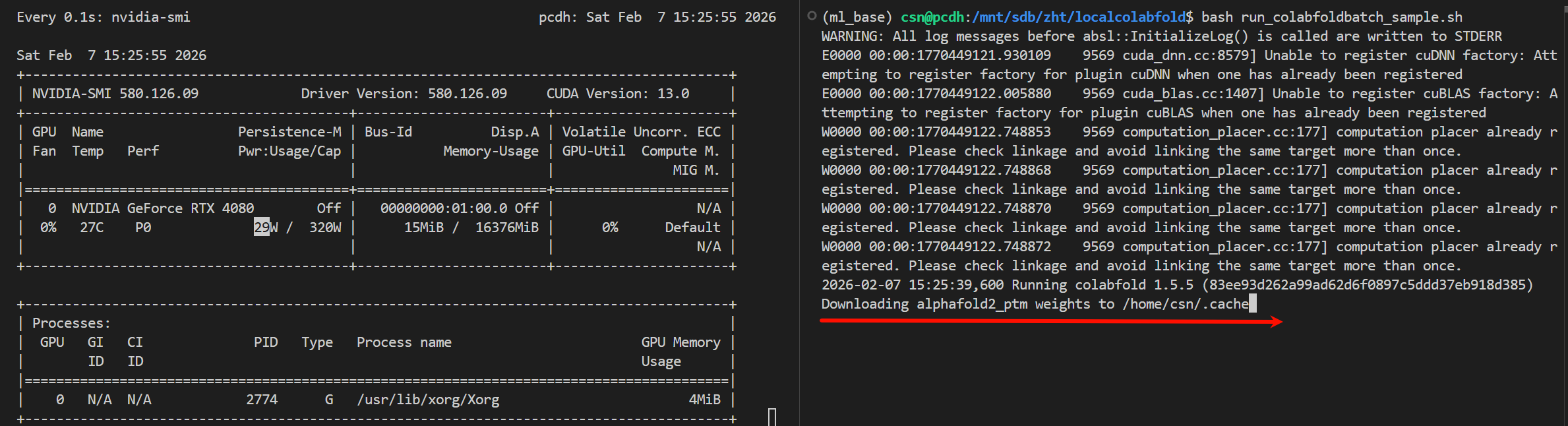
为了能够让大家更清楚地看到运行命令的时候发生了什么事情,我将屏幕上的标注输出流的提示信息,一并粘贴如下
markdown
WARNING: All log messages before absl::InitializeLog() is called are written to STDERR
E0000 00:00:1770449121.930109 9569 cuda_dnn.cc:8579] Unable to register cuDNN factory: Attempting to register factory for plugin cuDNN when one has already been registered
E0000 00:00:1770449122.005880 9569 cuda_blas.cc:1407] Unable to register cuBLAS factory: Attempting to register factory for plugin cuBLAS when one has already been registered
W0000 00:00:1770449122.748853 9569 computation_placer.cc:177] computation placer already registered. Please check linkage and avoid linking the same target more than once.
W0000 00:00:1770449122.748868 9569 computation_placer.cc:177] computation placer already registered. Please check linkage and avoid linking the same target more than once.
W0000 00:00:1770449122.748870 9569 computation_placer.cc:177] computation placer already registered. Please check linkage and avoid linking the same target more than once.
W0000 00:00:1770449122.748872 9569 computation_placer.cc:177] computation placer already registered. Please check linkage and avoid linking the same target more than once.
2026-02-07 15:25:39,600 Running colabfold 1.5.5 (83ee93d262a99ad62d6f0897c5ddd37eb918d385)
Downloading alphafold2_ptm weights to /home/csn/.cache
2026-02-07 15:29:26,425 Running on GPU
2026-02-07 15:29:29,479 Found 9 citations for tools or databases
2026-02-07 15:29:29,479 Query 1/1: 1BJP_1 (length 62)
COMPLETE: 100%|███████████████████████████████████| 150/150 [elapsed: 00:08 remaining: 00:00]
2026-02-07 15:29:44,206 Sequence 0 found templates: ['3mb2_C', '2fm7_B', '4fdx_A', '3ry0_B', '1bjp_A', '6fps_P', '4faz_C', '7m59_B', '6bgn_C', '1otf_D', '3abf_B', '5clo_C', '6fps_R', '7xuy_A', '5cln_I', '6blm_A', '7puo_F', '2op8_A', '4x1c_F', '6blm_A']
2026-02-07 15:29:44,605 Setting max_seq=512, max_extra_seq=5120
WARNING: All log messages before absl::InitializeLog() is called are written to STDERR
I0000 00:00:1770449390.232958 9569 mlir_graph_optimization_pass.cc:425] MLIR V1 optimization pass is not enabled
2026-02-07 15:30:23,616 alphafold2_ptm_model_1_seed_000 recycle=0 pLDDT=95.4 pTM=0.771
2026-02-07 15:30:43,596 alphafold2_ptm_model_1_seed_000 recycle=1 pLDDT=94.9 pTM=0.768 tol=0.248
2026-02-07 15:30:44,109 alphafold2_ptm_model_1_seed_000 recycle=2 pLDDT=94.9 pTM=0.766 tol=0.185
2026-02-07 15:30:44,623 alphafold2_ptm_model_1_seed_000 recycle=3 pLDDT=94.9 pTM=0.769 tol=0.334
2026-02-07 15:30:44,624 alphafold2_ptm_model_1_seed_000 took 53.8s (3 recycles)
2026-02-07 15:30:45,154 alphafold2_ptm_model_2_seed_000 recycle=0 pLDDT=95.1 pTM=0.773
2026-02-07 15:30:45,668 alphafold2_ptm_model_2_seed_000 recycle=1 pLDDT=94.8 pTM=0.771 tol=0.216
2026-02-07 15:30:46,182 alphafold2_ptm_model_2_seed_000 recycle=2 pLDDT=94.7 pTM=0.771 tol=0.352
2026-02-07 15:30:46,696 alphafold2_ptm_model_2_seed_000 recycle=3 pLDDT=94.7 pTM=0.771 tol=0.088
2026-02-07 15:30:46,696 alphafold2_ptm_model_2_seed_000 took 2.1s (3 recycles)
2026-02-07 15:31:03,536 alphafold2_ptm_model_3_seed_000 recycle=0 pLDDT=95.7 pTM=0.769
2026-02-07 15:31:18,938 alphafold2_ptm_model_3_seed_000 recycle=1 pLDDT=95.8 pTM=0.772 tol=0.259
2026-02-07 15:31:19,451 alphafold2_ptm_model_3_seed_000 recycle=2 pLDDT=95.8 pTM=0.776 tol=0.0578
2026-02-07 15:31:19,965 alphafold2_ptm_model_3_seed_000 recycle=3 pLDDT=95.9 pTM=0.775 tol=0.0903
2026-02-07 15:31:19,966 alphafold2_ptm_model_3_seed_000 took 33.3s (3 recycles)
2026-02-07 15:31:20,485 alphafold2_ptm_model_4_seed_000 recycle=0 pLDDT=95.9 pTM=0.767
2026-02-07 15:31:20,998 alphafold2_ptm_model_4_seed_000 recycle=1 pLDDT=95.7 pTM=0.771 tol=0.148
2026-02-07 15:31:21,512 alphafold2_ptm_model_4_seed_000 recycle=2 pLDDT=95.6 pTM=0.773 tol=0.103
2026-02-07 15:31:22,026 alphafold2_ptm_model_4_seed_000 recycle=3 pLDDT=95.5 pTM=0.773 tol=0.0478
2026-02-07 15:31:22,026 alphafold2_ptm_model_4_seed_000 took 2.1s (3 recycles)
2026-02-07 15:31:22,544 alphafold2_ptm_model_5_seed_000 recycle=0 pLDDT=95.9 pTM=0.775
2026-02-07 15:31:23,057 alphafold2_ptm_model_5_seed_000 recycle=1 pLDDT=95.2 pTM=0.771 tol=0.222
2026-02-07 15:31:23,571 alphafold2_ptm_model_5_seed_000 recycle=2 pLDDT=94.8 pTM=0.767 tol=0.173
2026-02-07 15:31:24,084 alphafold2_ptm_model_5_seed_000 recycle=3 pLDDT=94.8 pTM=0.766 tol=0.0928
2026-02-07 15:31:24,084 alphafold2_ptm_model_5_seed_000 took 2.1s (3 recycles)
2026-02-07 15:31:24,087 reranking models by 'plddt' metric
/mnt/sdb/zht/localcolabfold/.pixi/envs/default/lib/python3.12/site-packages/openmm/__init__.py:16: SyntaxWarning: invalid escape sequence '\p'
os.environ['PATH'] = '%(lib)s;%(lib)s\plugins;%(path)s' % {
/mnt/sdb/zht/localcolabfold/.pixi/envs/default/lib/python3.12/site-packages/openmm/openmm.py:11825: SyntaxWarning: invalid escape sequence '\S'
match = re.search("<([^?]\S*)", inputString)
/mnt/sdb/zht/localcolabfold/.pixi/envs/default/lib/python3.12/site-packages/openmm/app/internal/pdbstructure.py:1078: SyntaxWarning: invalid escape sequence '\.'
if not re.match("pdb.%2s.\.ent\.gz" % subdir , pdb_file):
/mnt/sdb/zht/localcolabfold/.pixi/envs/default/lib/python3.12/site-packages/openmm/app/internal/pdbstructure.py:1089: SyntaxWarning: invalid escape sequence '\.'
if not re.match("pdb.%2s.\.ent\.gz" % subdir , pdb_file):
/mnt/sdb/zht/localcolabfold/.pixi/envs/default/lib/python3.12/site-packages/openmm/app/internal/pdbx/reader/PdbxReader.py:347: SyntaxWarning: invalid escape sequence '\S'
"(?:_(.+?)[.](\S+))" "|" # _category.attribute
/mnt/sdb/zht/localcolabfold/.pixi/envs/default/lib/python3.12/site-packages/openmm/app/internal/pdbx/reader/PdbxReader.py:349: SyntaxWarning: invalid escape sequence '\s'
"(?:['](.*?)(?:[']\s|[']$))" "|" # single quoted strings
/mnt/sdb/zht/localcolabfold/.pixi/envs/default/lib/python3.12/site-packages/openmm/app/internal/pdbx/reader/PdbxReader.py:350: SyntaxWarning: invalid escape sequence '\s'
"(?:[\"](.*?)(?:[\"]\s|[\"]$))" "|" # double quoted strings
/mnt/sdb/zht/localcolabfold/.pixi/envs/default/lib/python3.12/site-packages/openmm/app/internal/pdbx/reader/PdbxReader.py:352: SyntaxWarning: invalid escape sequence '\s'
"(?:\s*#.*$)" "|" # comments (dumped)
/mnt/sdb/zht/localcolabfold/.pixi/envs/default/lib/python3.12/site-packages/openmm/app/internal/pdbx/reader/PdbxReader.py:354: SyntaxWarning: invalid escape sequence '\S'
"(\S+)" # unquoted words
/mnt/sdb/zht/localcolabfold/.pixi/envs/default/lib/python3.12/site-packages/openmm/app/internal/pdbx/reader/PdbxReader.py:419: SyntaxWarning: invalid escape sequence '\S'
"(?:_(.+?)[.](\S+))" "|" # _category.attribute
/mnt/sdb/zht/localcolabfold/.pixi/envs/default/lib/python3.12/site-packages/openmm/app/internal/pdbx/reader/PdbxReader.py:421: SyntaxWarning: invalid escape sequence '\s'
"(?:['\"](.*?)(?:['\"]\s|['\"]$))" "|" # quoted strings
/mnt/sdb/zht/localcolabfold/.pixi/envs/default/lib/python3.12/site-packages/openmm/app/internal/pdbx/reader/PdbxReader.py:423: SyntaxWarning: invalid escape sequence '\s'
"(?:\s*#.*$)" "|" # comments (dumped)
/mnt/sdb/zht/localcolabfold/.pixi/envs/default/lib/python3.12/site-packages/openmm/app/internal/pdbx/reader/PdbxReader.py:425: SyntaxWarning: invalid escape sequence '\S'
"(\S+)" # unquoted words
/mnt/sdb/zht/localcolabfold/.pixi/envs/default/lib/python3.12/site-packages/openmm/app/internal/amber_file_parser.py:65: SyntaxWarning: invalid escape sequence '\('
FORMAT_RE_PATTERN=re.compile("([0-9]+)\(?([a-zA-Z]+)([0-9]+)\.?([0-9]*)\)?")
/mnt/sdb/zht/localcolabfold/.pixi/envs/default/lib/python3.12/site-packages/openmm/app/gromacsgrofile.py:62: SyntaxWarning: invalid escape sequence '\.'
return match('^[-+]?[0-9]*\.?[0-9]*([eEdD][-+]?[0-9]+)?$',word)
/mnt/sdb/zht/localcolabfold/.pixi/envs/default/lib/python3.12/site-packages/openmm/app/metadynamics.py:247: SyntaxWarning: invalid escape sequence '\.'
pattern = re.compile('bias_(.*)_(.*)\.npy')
2026-02-07 15:31:31,002 Relaxation took 6.9s
2026-02-07 15:31:31,003 rank_001_alphafold2_ptm_model_3_seed_000 pLDDT=95.9 pTM=0.775
2026-02-07 15:31:32,437 Relaxation took 1.4s
2026-02-07 15:31:32,437 rank_002_alphafold2_ptm_model_4_seed_000 pLDDT=95.5 pTM=0.773
2026-02-07 15:31:33,375 Relaxation took 0.9s
2026-02-07 15:31:33,375 rank_003_alphafold2_ptm_model_1_seed_000 pLDDT=94.9 pTM=0.769
2026-02-07 15:31:34,757 Relaxation took 1.4s
2026-02-07 15:31:34,757 rank_004_alphafold2_ptm_model_5_seed_000 pLDDT=94.8 pTM=0.766
2026-02-07 15:31:35,711 Relaxation took 1.0s
2026-02-07 15:31:35,712 rank_005_alphafold2_ptm_model_2_seed_000 pLDDT=94.7 pTM=0.771
2026-02-07 15:31:36,016 Done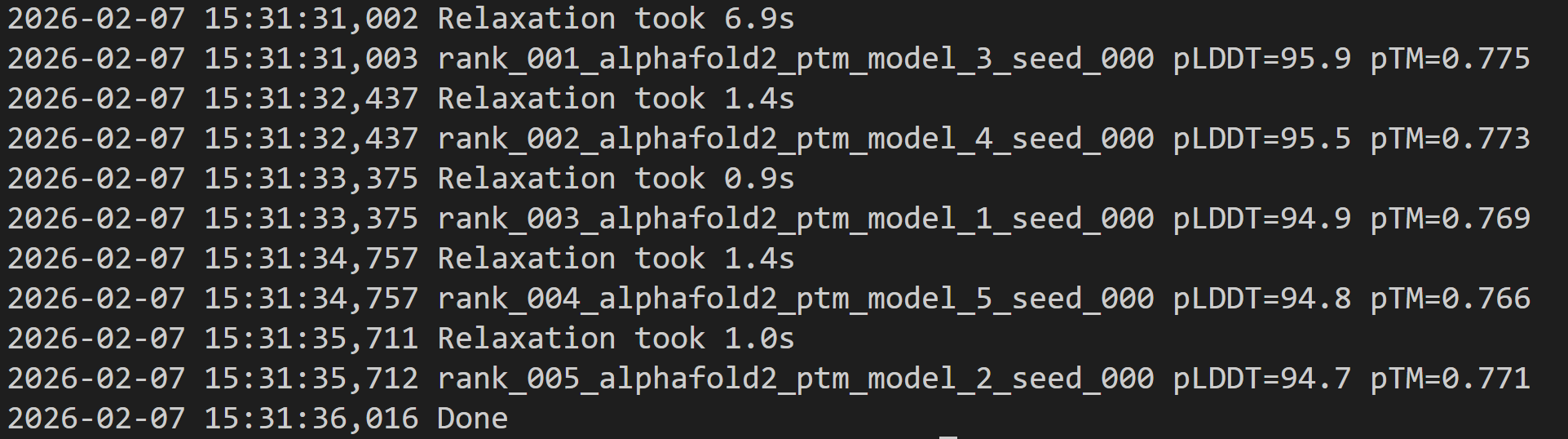
其实我们可以发现,输出的5个model已经自己为我们排好序了,rank1的model_3是最好的。
当然,这里的rank注释方式和我们在alphafold3中的model_0那种默认方式不同,还是需要注意一下的。
下面是整个输出文件夹:
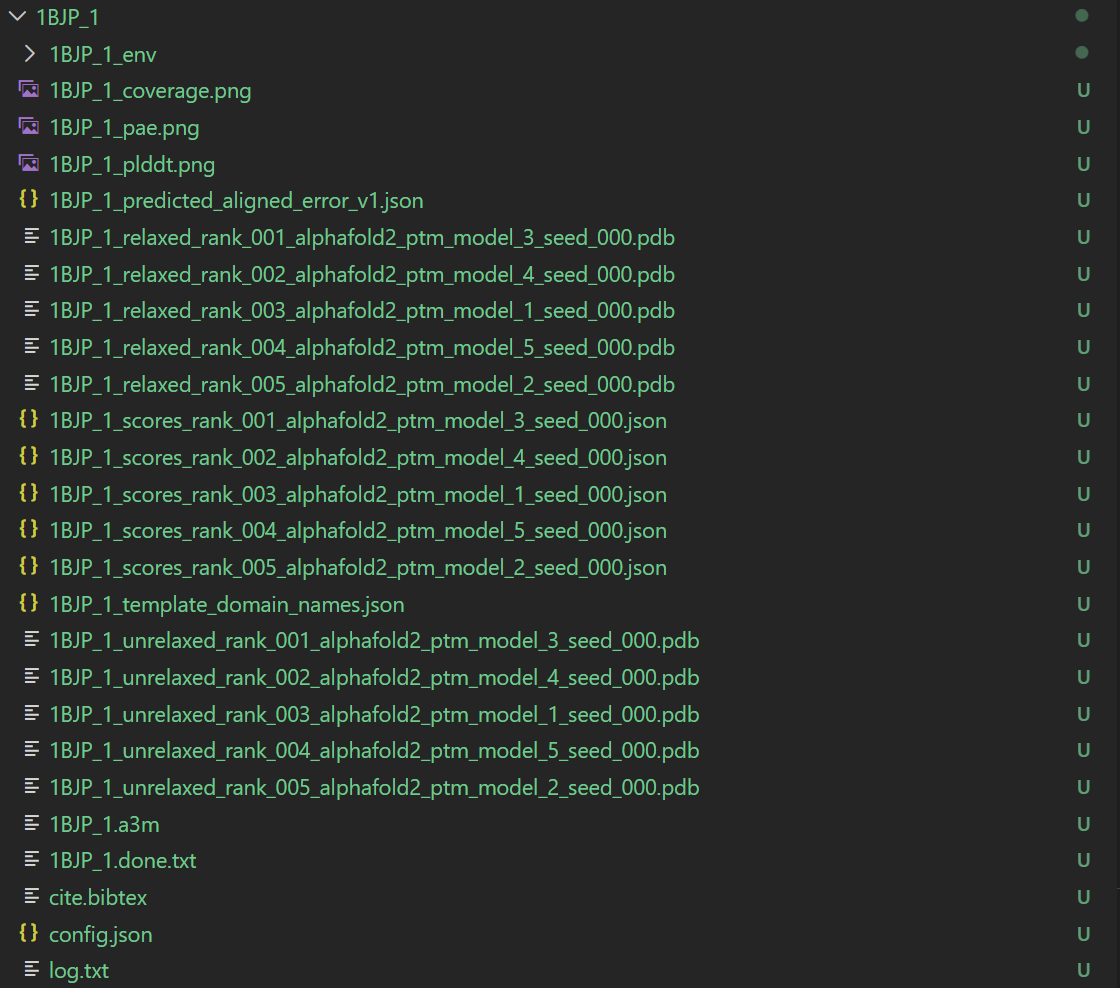
6,脚本示例输出结果解读
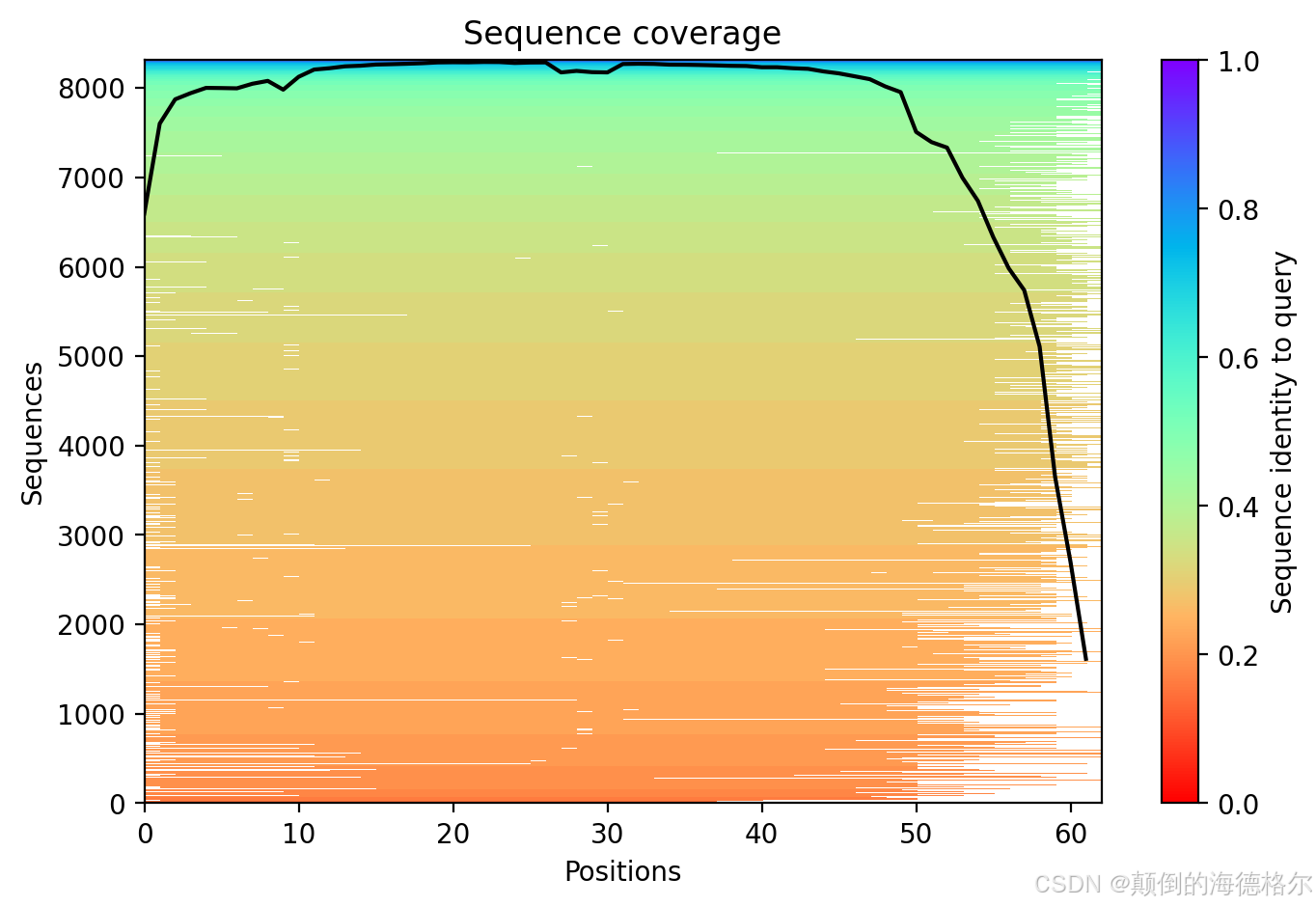
然后接着是PAE矩阵
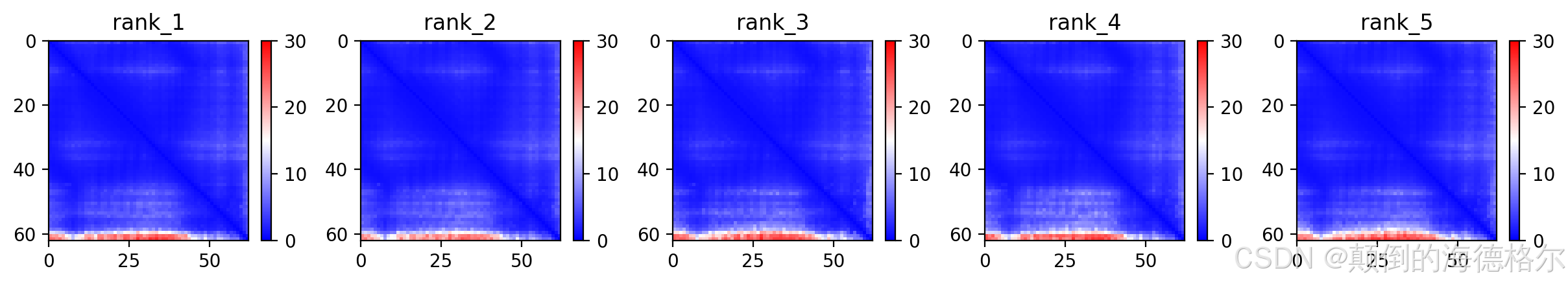
紧接着是PLDDT指标,
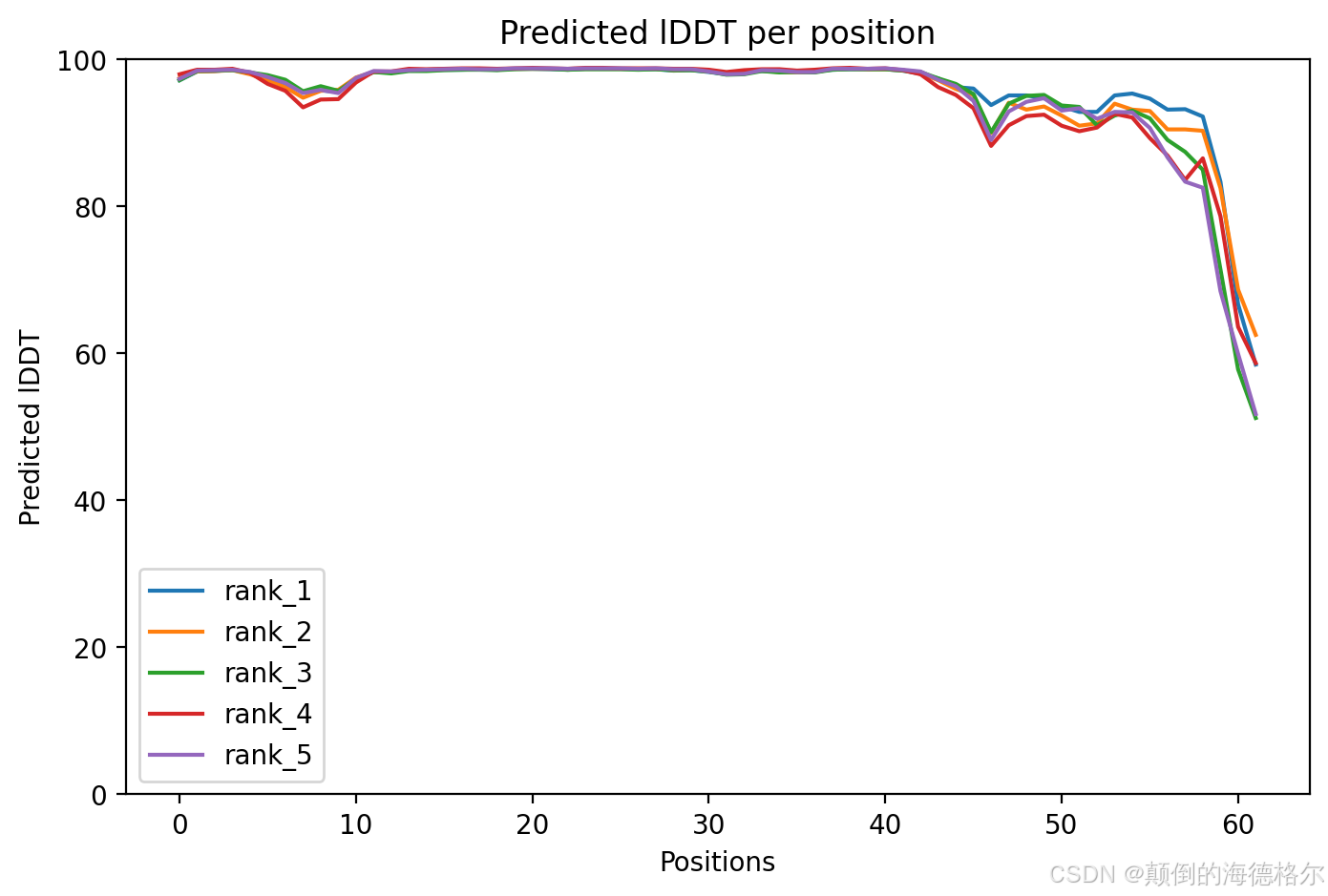
- 结构文件.pdb:
unrelaxed:原始坐标,原子之间可能存在微小的物理冲突;
relaxed:经过OpenMM的 Amber力场能量最小化,微调了侧链位置,消除了原子重叠,使结构在物理化学上更合理。后续做分子对接(Docking)或分子动力学模拟、互作分析必须用这个版本 - Metric文件.json:plddt、PAE
- 多序列比对.a3m:进化信息
7,其他的演示例子
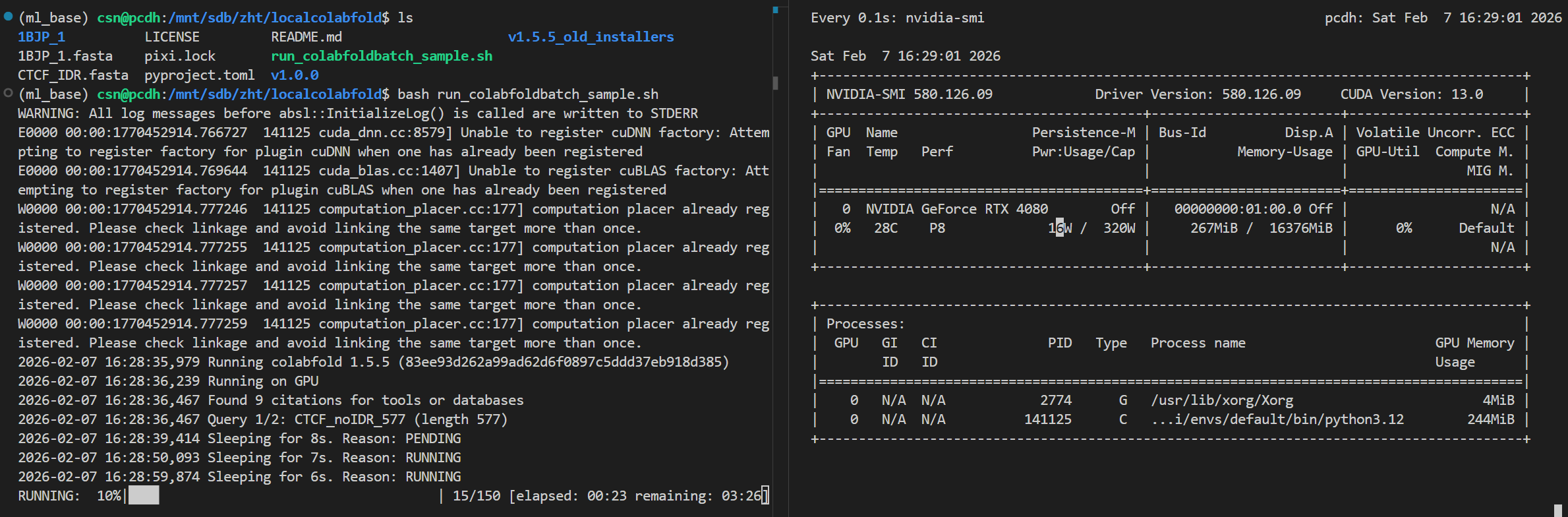
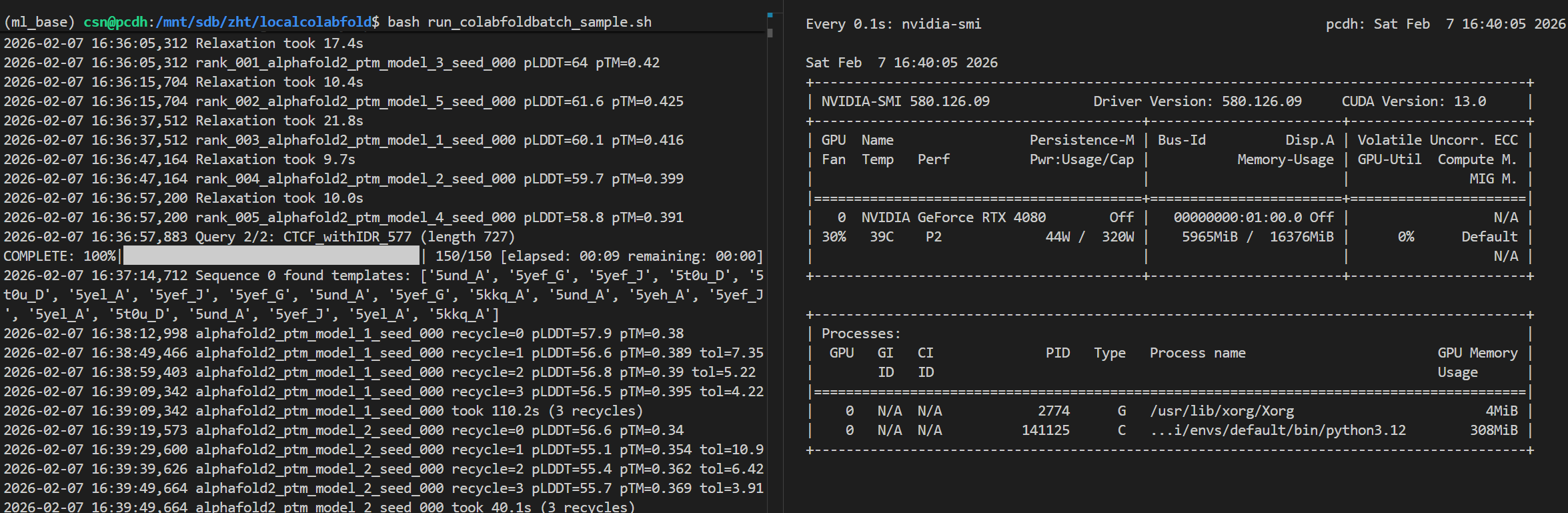
我此处输入了两个蛋白质序列,在1个多序列fasta文件中,一起处理。
大概可以看到,1个批次处理2个样本数据时,然后序列长度大概在500aa左右的fasta数据时,
显存大概会干到6G
这里,我们一般的需求是想进一步查看到底预测出来的结构model如何,也就是可视化,
但是导入到在线工具、pymol奇美拉之类的分子可视化软件毕竟是挺麻烦的,
虽然很多人下意识都是这么做,
其中关于开源pymol可视化,可以参考我之前的博客:
使用AlphaFold3预测蛋白质三维结构及PyMol可视化
我在这里的需求是直接在notebook中可视化三维结构,此处介绍一个我很久前用的可视化插件------ py3Dmol。
py3Dmol将是在Jupyter上运行3Dmol.js的小部件。并非所有功能都可以使用,但是许多API是通用的,因此阅读3Dmol.js官方网站是学习的最佳方法