导言
论文地址:https://arxiv.org/abs/2208.04314
深度学习在免疫学领域的应用
人们有一种叫做免疫系统 的系统来保护自己免受外来有害物质(包括病毒和细菌)的侵害。免疫系统是指包括白细胞在内的人体内各种元素相互配合以保卫身体的系统,众所周知,它是人类生存的重要机制。近年来,人们一直在研究如何利用免疫系统的机制来治疗难以治愈的癌症。
免疫系统中一个特别重要的组成部分是HLA 分子,它通过向另一个细胞呈现一种叫做肽的物质来诱导免疫反应,这种物质会被一个细胞吸收,因此它是发生免疫反应的一个重要组成部分。近年来,人们一直在进行研究,以阐明免疫系统的机制,并了解 HLA 如何呈现肽。
特别是,HLA 可根据其组成单元的序列分为多个版本(这些不同的基因版本称为等位基因)。根据这些分类准确预测肽的表现形式是一项重要的临床挑战。
在本文中,除了使用一般数据集进行实验外,还利用了黑色素瘤(一种皮肤癌)细胞的数据进行实验,证明了该模型在临床应用方面的潜力。
当前工具和研究流的局限性和问题
在过去二十年中,人们开发了许多工具来预测 HLA 肽的结合。特别是近年来,利用深度学习 的模型得到了广泛应用。然而,这些模型仅适用于有限数量的 HLA 等位基因(版本),在实际准确性方面存在不足(已知 HLA 可分为 HLA-I 和 HLA-II,HLA-II 的这一趋势尤为明显)。
众所周知,当与 HLA 结合的肽具有一定长度(如 9 或 10)时,预测准确率较高,但当肽较长时,由于缺乏 具有该长度的训练样本 ,预测性能会明显降低。另一个挑战是,目前的方法没有充分利用数据 (尤其是蛋白质之间的序列上下文信息)与生物信息 之间的关系。
因此,本文提出了TripHLApan来解决这些问题。
型号详情
整体模型

TripHLApan的整体工作流程如图 a 所示。
在 TripHLApan 模型中,肽序列和 HLA 分子均来自 IEDB 数据库,并以字符串的形式表示为输入数据(如图所示,每个 HLA 和肽构件均用一个英文字母表示)。这些输入数据在训练前已根据各种属性对 HLA 分子和肽进行了预处理。
需要注意的是,在本实验中,首先要对数据进行选择,使训练集、测试数据集和包含不包含在训练数据中的等位基因的数据集(以下定义为未见数据集)不相互重叠。
上述作为输入的数据采用了三种编码方法 :AAIndex、Blosum62 和 Embedding。通过这三种编码方法的并行****化 过程,可以获得潜在的多方面信息,如****生物化学特性和 结合的物理信息,而这些信息是无法仅从表面的序列信息中看到的。
编码模型的输出被用作名为 BiGRU的模型的输入。
此外,本文中的模型还使用了 BiGRU 模型中的注意力机制,以反映序列中哪些是学习的重要点(下文将讨论在该模型中使用 BiGRU 模块和注意力机制的原因)。
由此获得的三个矩阵合并后输出(在最终输出前使用全合并层或西格码层)。该模型表明,利用这种并行多重编码方法进行学习,可以从多个角度利用氨基酸的特性。
BiGRU 模型的详情以及使用该模型的原因
BiGRU(双向门控递归单元)模型是RNN 模型的扩展;BiGRU 的最重要特点之一是,它们涉及阵列正反 两个方向的信息处理过程。
与只从一个方向进行训练的普通 RNN 模型不同,字符串是从正反两个方向进行训练的,这样就能更好地捕捉字符序列的上下文信息。
BiGRU 还引入了一种门控机制来捕捉长期依赖关系。此外,在 TripHLApan 中,BiGRU 模型还增加了注意机制:注意机制包括一个根据序列的重要性重新分配权重的过程,从而使其能够充分反映上下文所包含的信息。
因此,通过利用 BiGRU 和 Attention 机制,即使三维结构不足以预测 HLA 和多肽,也可以最大限度地利用序列上下文信息进行学习。在本文中,该模型的最大优势之一是能够了解直接与 HLA 结合的多肽末端是如何影响结合的。
迁移学习简介
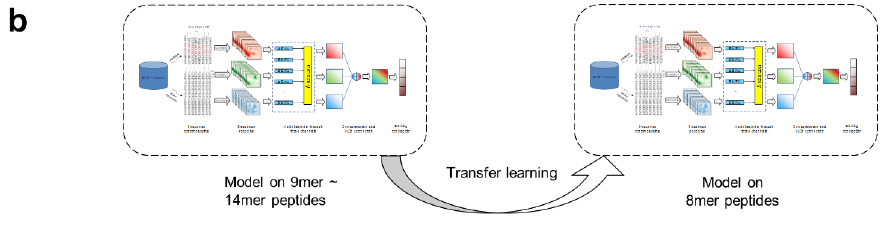
如图 b 所示,该模型还引入了过渡学习 ,以解决因缺乏 长度较长的肽的训练数据而导致的预测准确性不高的问题。引入这种过渡学习的原因之一是,当肽的长度为 8 时,已知会出现一种特殊的耦合。
因此,该机制是这样的:在训练的早期阶段,利用长度在 9 到 14 之间的多肽(即长度相对较长的多肽数据)对模型进行训练,然后利用训练得到的模型对长度为 8 的多肽进行预测。这种机制使得在预测长度大于 8 的多肽时,学习不受长度为 8 的数据的影响,并防止过度拟合特定多肽长度的数据。
实验结果
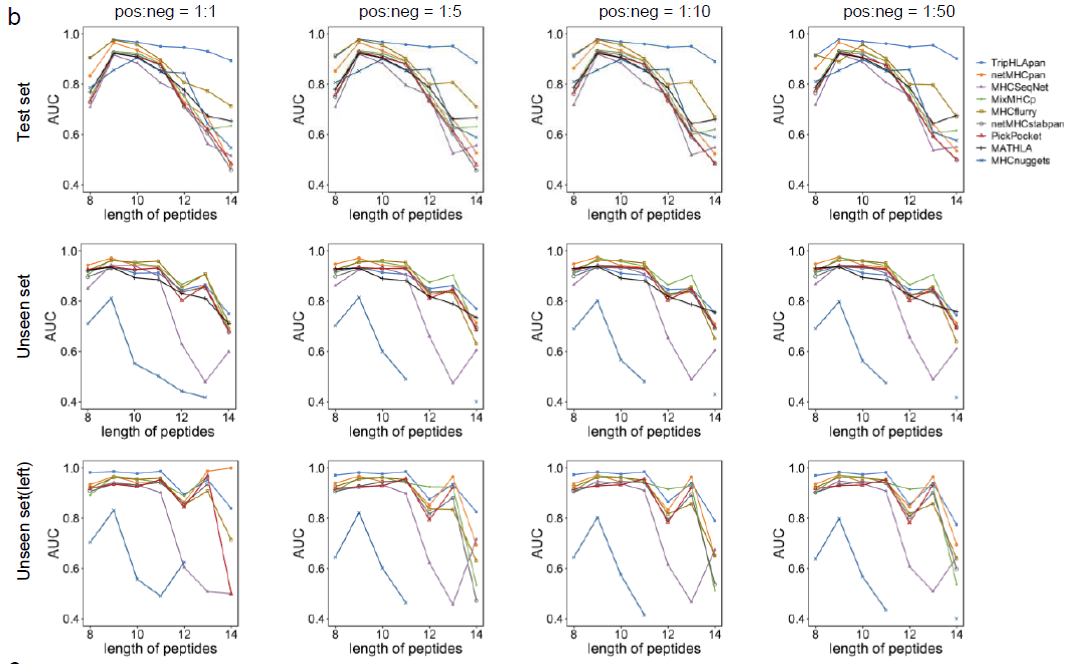
在图 b 中,显示了在不同的阳性样本和阴性样本比例下(具体来说,左起四幅图中阳性样本和阴性样本的比例分别为 1:1、1:5、1:10 和 1:50)测量 BiGRUAUC 的实验结果。横轴表示实验中使用的肽段长度(在本实验中,学习是在一定程度上根据肽段长度进行分类的)。
图 b 包括三行:最上面一行显示的是测试集上的 AUC,中间一行显示的是使用未见数据 集(如上所述,数据集包含未包含在训练数据中的等位基因(版本))时的 AUC,最下面一行显示的是使用未见数据集和迁移学习时的 AUC。最下面一行显示的是使用未见数据集进行迁移学习时的 AUC。图中蓝色部分显示的是本文提出的模型的评价指数,其他彩色部分显示的是传统模型的评价指数。
从图中可以看出,新方法在所有肽段长度上的表现都优于传统方法,尤其是在较长的肽段长度上。下图还显示了过渡学习的有效性。
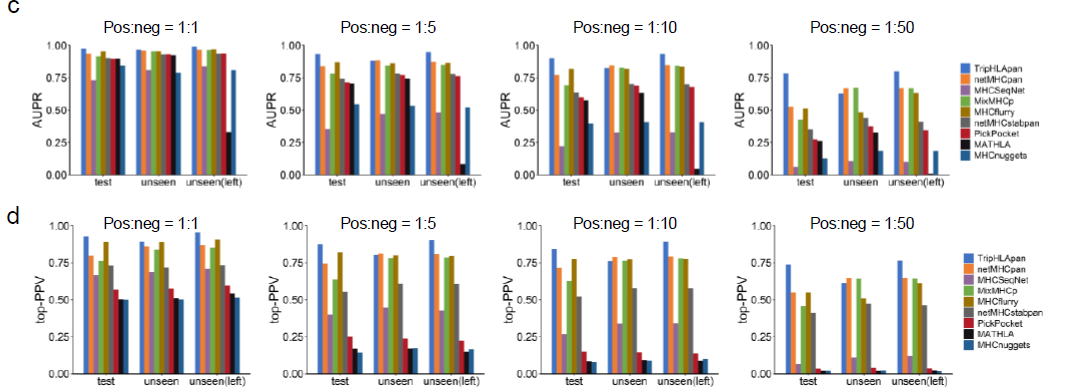
此外,图 c 和 d 显示了AUPR 和top-PPV,这是模型在使用不平衡数据集时的性能指标。这证实了该模型在数据不平衡情况下的有效性。
实验结果
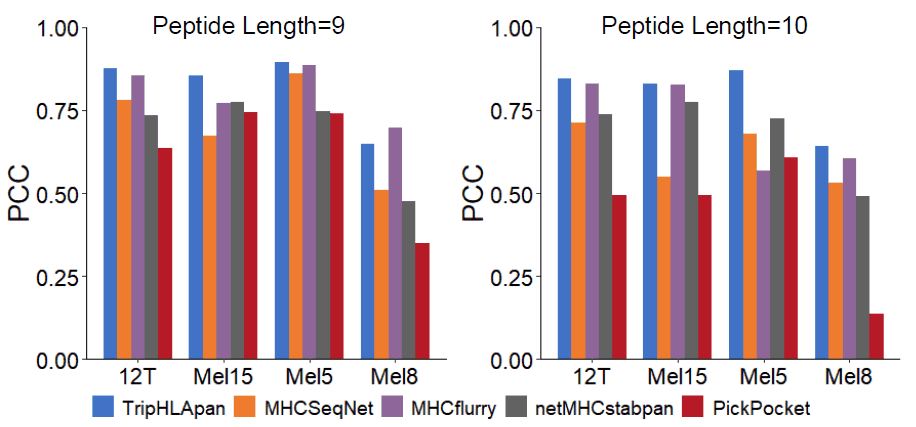
上图显示了使用预测工具对不同等位基因样本进行皮尔逊相关性测量 的结果,并在与单个黑色素瘤****相关的数据集上进行了测试(黑色素瘤是皮肤癌的一种,目前正在考虑引入免疫疗法)。获得的**平均皮尔逊相关系数 (PCC)**被用作纵轴,实验中使用的细胞系都与黑色素瘤有关(细胞系是为研究目的而持续培养的一组细胞)。
PCC 是用来衡量预测的肽-HLA 结合频率与实际结合频率相关性的指标。本文发现 TripHLApan 在所有肽长度和样本中都表现出较高的 PCC。
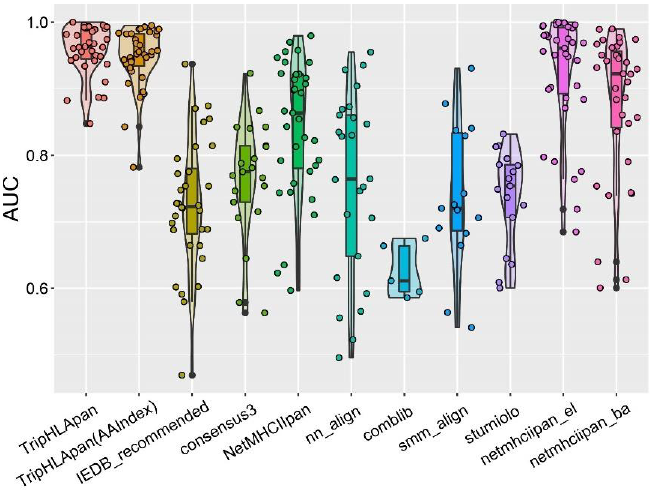
众所周知,HLA 可根据其功能分为 I 和 II 两类。之前的实验表明,HLA-I 的性能很高,但如上图所示,TripHLApan 的模型对 II 也显示出了出色的 AUC 值。这表明该模型可能对 HLA-II 特别有效,因为它之前只在有限的数据集上进行了验证,确保了足够的预测准确性。
总结
与传统方法相比,HLA 分子和肽的准确性更高,这是因为在利用分子的生物和统计特性、BiGRU 架构 和注意力模块 的组合以及迁移学习进行适当预处理后,整合了多种信息和并行编码。结果表明,与传统方法相比,准确度有所提高。这可能是因为能够从多个角度利用生物特性和序列上下文信息。
在 HLA-I 模型与 HLA-II 模型的比较中,TripHLApan 的表现优于 目前最先进的预测工具 ,无论是在一般数据集还是在与皮肤癌细胞、黑色素瘤相关的数据集中。
未来面临的一个挑战是,对于肽段长度为 9 的样本,HLA-I 与肽段的结合预测还没有足够的改进,而这是 HLA-I 结合预测中最常见的肽段长度。因此,今后必须设法更多地关注三维结构 ,因为目前还没有利用三维结构进行学习。就我个人而言,我认为多功能性非常重要,它可以让过渡学习的肽长度设置更加灵活,而不仅仅是将其设置为一个预先指定的值(本例中为 8)。